Основность кислот — Справочник химика 21
Эквивалентную массу вещества вычисляют, исходя из его мольной массы. Эквивалентная масса кислоты равна ее мольной массе, деленной на основность кислоты. Эквивалентная масса основания равна его мольной массе, деленной на валентность металла, образующего основание. Эквивалентная масса [c.44]Основность кислоты определяется числом протонов, которое отдает молекула кислоты, реагируя с основанием кислотность основания определяется числом протонов, присоединяемых молекулой основания прн взаимодействии его с кислотой. [c.8]
Соли двух- и более основных кислот так же, как и многоосновных оснований, гидролизуются ступенчато, например [c.65]
Основностью КИСЛОТЫ называется число атомов водорода в мо лекуле кислсггы, способных замещаться нз металл с образованием соли. Такие кислоты, как соляная и уксусная, могут служить при-мерам н одноосновных кислот, серная кислота двухосновна, орто-фосфО()ная кислота Н3РО4 трехосновна.
В нефтях может содержаться значительное количество кислот. Основные кислоты нафтеновые. Их содержание иногда достигает 3% (нафталанская лечебная нефть). Меньше содержится ароматических, нафтено-ароматических и жирных кислот [8]. [c.11]
На нейтрализацию 4 молей кислоты израсходовано 3 г-экв едкого натра. Чему равна основность кислоты [c.89]
Исследуя кислоты, полученные при окислении парафиновых углеводородов изостроения, можно составить представление о пунктах окислительной атаки кислорода. Последний действует преимущественно на точку разветвления, иначе говоря, на третичный атом водорода, В результате отщепления боковых цепей образуются в основном кислоты с прямой цепью. Тем не менее парафины с сильно разветвленным угле- родным скелетом продолжают оставаться непонгодными для промышленных целей сырьем [42], При их окислении получают главным обраэом низкомолекулярные и более глубоко окисленные карбоновые кислоты с числом атомов углерода меньше 12, не говоря уже о значительных количествах кислот с разветвленным скелетом.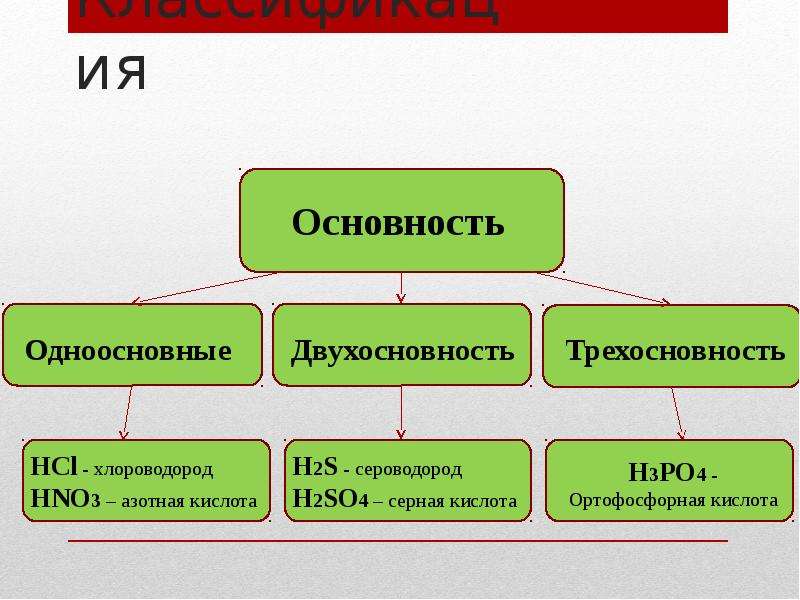
Молярную массу эквивалентов вещест.ва вычисляют, исходя из его молярной массы. Молярная масса эквивалентов кислоты равна ее молярной массе, деленной на основность кислоты. Молярная масса эквивалентов основания равна его молярной массе, деленной на валентность металла, образующего основание. Молярная масса эквивалентов соли равна ее молярной массе, деленной на произведение валентности металла на число его атомов в молекуле.
Известен целый ряд полиамидов, отличающихся по строению исходных мономеров. Первым полиамидом, из которого стали делать синтетические волокна, был нейлон-6,6 называемый также анид. Этот полиамид был получен при исследованиях Карозерса в 1935 г. из гексаметилендиамина и адипиновой кислоты. Известны и другие виды нейлона, получаемые на основе иных диаминов и двух основных кислот — нейлон-6,10, нейлон-11 и др. [c.348]
из гексаметилендиамина и адипиновой кислоты. Известны и другие виды нейлона, получаемые на основе иных диаминов и двух основных кислот — нейлон-6,10, нейлон-11 и др. [c.348]
Свойства ортоборной кислоты. 1. Приготовьте несколько миллилитров насыщенного при комнатной температуре раствора ортоборной кислоты и определите его pH при помощи универсального индикатора. Какова основность кислоты Напишите уравнение диссоциации. [c.235]
Сила кислот связана с их основностью. Чем выше основность кислот, определяемая числом ионов водорода в молекуле кислоты, способных замещаться металлом, тем меньше сила кислоты. Трех- и четырехосновные кислоты -не бывают сильными. [c.174]
Соли образуются за счет замещения всех или части атомов водорода кислоты атомами металла. Если исходная кислота является двух и более основной, то атомами металла могут быть замещены не все атомы водорода. В результате валентность кислотного остатка окажется меньше основности кислоты.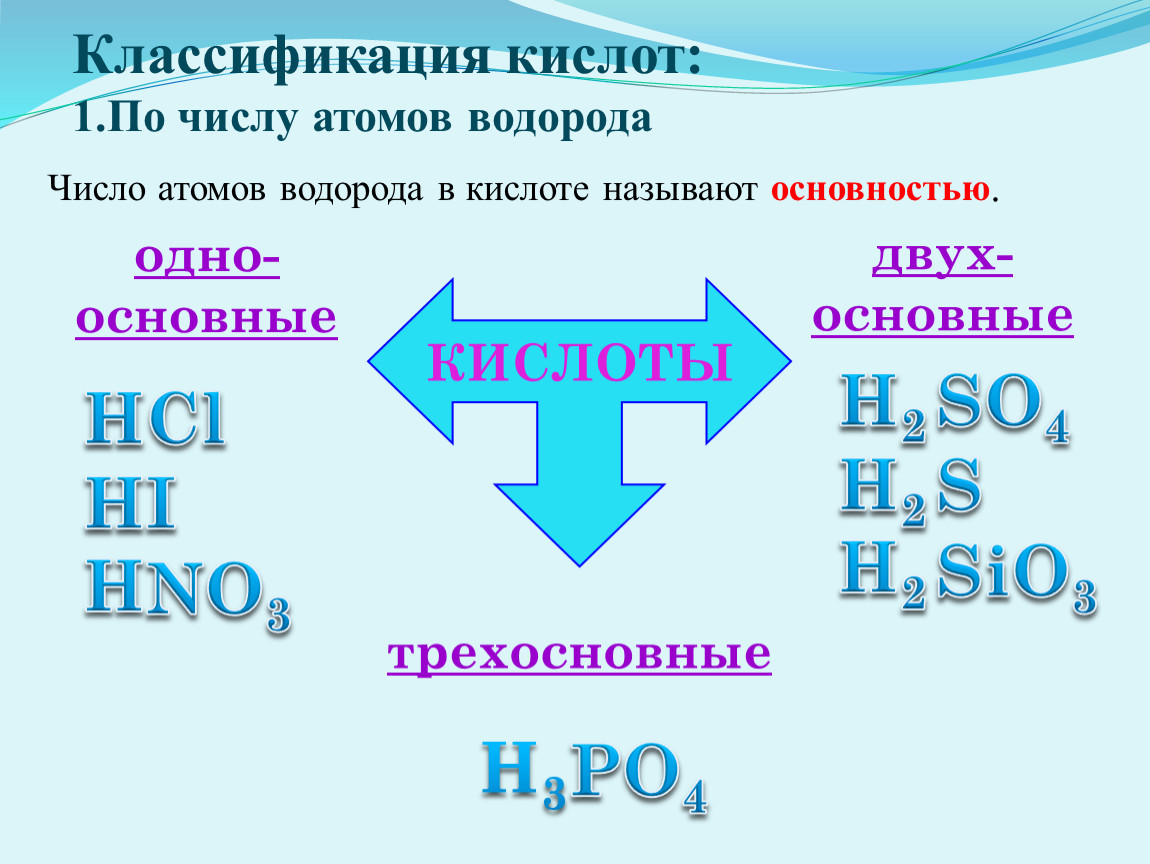 Валентность кислотного остатка определяется как число атомов водорода исходной кислоты, которые были замещены атомами металла. Например, Н80 , остаток серной кислоты Н 80 , будет одновалентным, поскольку ему не хватает одного атома водорода. [c.13]
Валентность кислотного остатка определяется как число атомов водорода исходной кислоты, которые были замещены атомами металла. Например, Н80 , остаток серной кислоты Н 80 , будет одновалентным, поскольку ему не хватает одного атома водорода. [c.13]
В общем случае п-основной кислоты получаются уравнения подобного же типа, знаменатель которых состоит из и+1 слагаемых. Так, для трехосновной кислоты НзА знаменатель этих уравнений содержит четыре слагаемых [Н» [Н ] + К К2 [Н ] -Ь 1 2/Сз, а при вычислении долей НзА, НгА , НА и А каждое из слагаемых поочередно выполняет функцию числителя, например
Наивысшая основность кислоты достигается в большинстве солей. [c.227]
Кислоты кислородные и бескислородные 72, 98, 100—101 Основность кислот 74, [c.187]
Измерьте pH 1 М раствора серной кислоты. Разбавлением в 10 раз приготовьте 0,1 М 0,01 М и т. д. растворы. На основании значений pH растворов сформулируйте выводы об основности кислоты, ее силе и изменении свойств при уменьшении концентрации. [c.232]
[c.232]
В обменных реакциях молярная масса эквивалента кислоты равна ее молярной массе, деленной на основность кислоты (число ионов водорода, замешенных металлом в реакции). Например [c.184]
Основность кислот — это число атомов водорода в молекуле кислоты, которые способны замещаться на металл с образованием соли, [c.17]
Число способных к ионизации атомов водорода определяет основность кислоты. [c.172]
Остаток после отгона бензойной кислоты содержал двухосновные кислоты, для разделения которых пользовались методом Тауша—Добрянского [9]. Из двух основных кислот в преобладающем количестве была обнаружена изофталсвая кислота из нее был получен ее диметиловый эфир, который после двухкратной перекристаллизации плавился при 65-67°. [c.26]
Экспериментально было установлено,,что при окислении в жидкой фазе высших парафинов в определенных условиях образуется много (около 50%) жирных кислот, которые содержат в среднем вдвое меньше атомов углерода, чем углеводороды сырья.
Как видно, Кг2 Кг1. Это связано с тем, что константа диссоциации кислоты по первой ступени, как правило, значитеаьно больше константы диссоциации по второй ступени. Поэтому при приближенных расчетах, связанных с гидролизом солей слабых много-основных кислот, можно принимать во внимание только гидролиз по первой ступени. [c.151]
Эквиваленты кислот H I, HNO3, СН3СООН составляют соответственно 36, 46, 63,01 и 60,03 ед. м., поскольку в них содержится гю 1,008 ед. м. водорода, замещающегося металлом. Именно поэтому эквивалент, кислоты может быть вычислен как частное от деления ее молекулярной массы на основность кислоты.
Сырье и возвратные парафины смешиваются с водным раствором борной кис-лоты в смесителенагреваются до 160 °С и закачиваются в реактор 2. Оксидат, пройдя подогреватель, с температурой 260 °С направляется в ректификационную колонну 3, в которой под вакуумом (остаточное давление 2,7 кПа, т. е. 20 мм рт. ст.) от борных эфиров отгоняются соединения, не вступившие в реакцию с борной кислотой (в основном кислоты, кетоны и непрореагировавшие углеводороды). Дистиллят после охлаждения поступает в омылитель 4, где он обрабатывается 10%-ным раствором едкого натра. В ходе омыления из отгона извлекаются все кислоты и протекает процесс частичного омыления соединений сложноэфирного характера. Омыленный продукт в отстойнике 5 разделяется на два слоя. Верхний, состоящий в основном из углеводородов, направляется в смеситель /. Мыльный
В отличие от кислородсодержащих соединений нефти, которые представлены в основном кислотами и фенолами, легко удаляемыми из нефтяных фракций щелочью, удалить сернистые соединения очень сложно. Это связано с тем, что большинство сернистых соединений нейтральны и очень близки по снойствамк ароматическим соединениям нефти. Даже меркаптаны, имеющие слабокислые свойства, по мере увеличения молекулярной массы теряют эти свойства и их выделение из нефтяных фракций с помощью п1елочи становится нецелесообразным. Все существующие в лабораторной и промышленной практике химические и физико-химические методы разделения — такие, как сульфирование, адсорбционная хроматография, экстракция, разделение с помощью комплексообразова-ния и ректификация — оказываются малоэффективными и пока неприемлемы для промышленности. [c.199]
Это связано с тем, что большинство сернистых соединений нейтральны и очень близки по снойствамк ароматическим соединениям нефти. Даже меркаптаны, имеющие слабокислые свойства, по мере увеличения молекулярной массы теряют эти свойства и их выделение из нефтяных фракций с помощью п1елочи становится нецелесообразным. Все существующие в лабораторной и промышленной практике химические и физико-химические методы разделения — такие, как сульфирование, адсорбционная хроматография, экстракция, разделение с помощью комплексообразова-ния и ректификация — оказываются малоэффективными и пока неприемлемы для промышленности. [c.199]
На нейтрализацию 0,943 г фосфористой кислоты НцРОз израсходовано 10 мл 2,3 н. раствора шелочи. Определите основность кислоты и напишите структурггую формулу. Почему не указаьга природа щелочи [c.35]
В зависимости от природы радикала, связанного с карбоксильной группой, карбоновые кислоты делятся на предельные и непредельные. Число карбоксильных групп определяет основность кислот кислоты с одной карбоксильной группой являются одноосновными, а с двумя — двухосновными и т. д. Кроме того, по количеству углеродных атомов в радикале различают кислоты низихие (низкомолекулярные) и высшие (высокомолекулярные). [c.140]
Число карбоксильных групп определяет основность кислот кислоты с одной карбоксильной группой являются одноосновными, а с двумя — двухосновными и т. д. Кроме того, по количеству углеродных атомов в радикале различают кислоты низихие (низкомолекулярные) и высшие (высокомолекулярные). [c.140]
Ионы водорода присутствуют во всех растворах кислот и придают им характерные свойства (кислый вкус, способность изменять окраску индикаторов, способность замещать протон катионами металлов при химических реакциях). Молекулы кислот могут содержать разное количество атомов водорода. Число атомов водорода определяет основность кислоты. С ществуют оМногоосновные кислоты диссоциируют ступенчато
Были сделаны попытки синтеза таких соединеяий. Основность кислот изучалась методами потенциометрического, кондуктометрического, термометрического титрования. Однако с помощью этих методов не удалось доказать правильность или отвергнуть формулы типа Н7[Р (М02О7) б]. Оказалось, что процессы замещения внешнесферных ионов гетерополисоединений сопровождаются сложными превращениями, связанными с разрушением внутренней сферы комплекса. [c.228]
Оказалось, что процессы замещения внешнесферных ионов гетерополисоединений сопровождаются сложными превращениями, связанными с разрушением внутренней сферы комплекса. [c.228]
Однако и в этом случае атом водорода, определяющий основность кислоты, связан с атомом неметалла через кислород. В неорга-нической химии таких примеров почти нет (упомянем только фосфорноватистую Н3РО2 и фосфористую Н3РО3 кислоты) [c.237]
Химия для поступающих в вузы 1985 (1985) — [ c.109 ]
Химия для поступающих в вузы 1993 (1993) — [ c.125 ]
Органическая химия (1968) — [ c.140 ]
Органическая химия 1965г (1965) — [ c.169 ]
Органическая химия 1969г (1969) — [ c.188 ]
Органическая химия 1973г (1973) — [
c.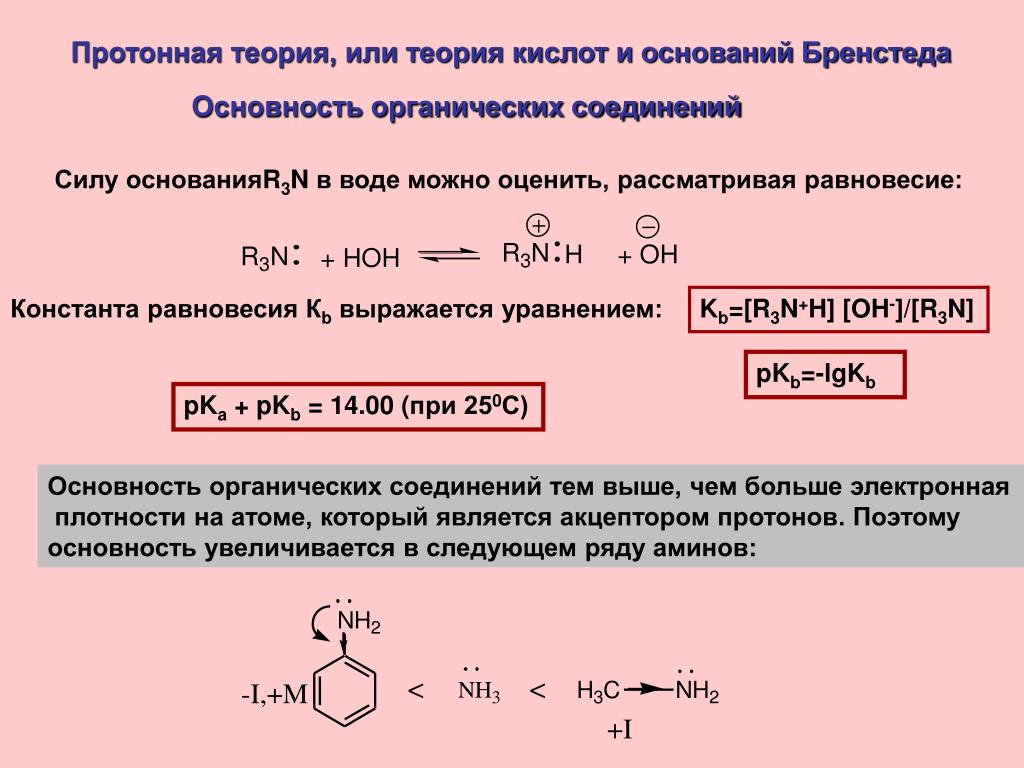 179
]
179
]
Неорганическая химия (1950) — [ c.98 ]
Органическая химия Издание 4 (1981) — [ c.176 ]
Органическая химия 1971 (1971) — [ c.142 ]
Органическая химия 1974 (1974) — [ c.117 ]
Основы химии Том 2 (1906) — [ c.570 ]
Химия (1985) — [ c.94 ]
Неорганическая химия (1950) — [ c.86 ]
Очерк общей истории химии (1979) — [ c.246 , c.304 ]
Химия (1982) — [ c.74 ]
Органическая химия Издание 6 (1972) — [ c.117 ]
Курс органической химии Издание 4 (1985) — [
c. 167
,
c.210
,
c.315
,
c.409
]
167
,
c.210
,
c.315
,
c.409
]
Химия Издание 2 (1988) — [ c.101 ]
Основы общей химии Т 1 (1965) — [ c.56 , c.174 ]
Основы общей химии Том 2 Издание 3 (1973) — [ c.55 , c.172 ]
Основы общей химии том №1 (1965) — [ c.56 , c.174 ]
Кислоты и основания • Джеймс Трефил, энциклопедия «Двести законов мироздания»
Из нашего повседневного опыта мы знаем, что некоторые вещества обладают высококоррозионными свойствами. Например, если кислота из аккумулятора вашей машины попадет на одежду, она сразу же ее проест. Иногда мы используем аммиак и другие вещества для домашней уборки. Эти коррозионные вещества известны химикам как кислоты и основания. На поверхностном уровне их различить совсем не сложно. Кислоты кислые на вкус и окрашивают лакмусовую бумажку в красный цвет, щелочи же мыльные на ощупь и окрашивают лакмусовую бумажку в синий цвет. Однако химики редко довольствуются такого рода феноменологическими определениями. Они ищут ответ на вопрос «Что делает вещество кислотой или основанием на молекулярном уровне?». Вот уже больше века химики бьются над определением кислот и оснований.
Иногда мы используем аммиак и другие вещества для домашней уборки. Эти коррозионные вещества известны химикам как кислоты и основания. На поверхностном уровне их различить совсем не сложно. Кислоты кислые на вкус и окрашивают лакмусовую бумажку в красный цвет, щелочи же мыльные на ощупь и окрашивают лакмусовую бумажку в синий цвет. Однако химики редко довольствуются такого рода феноменологическими определениями. Они ищут ответ на вопрос «Что делает вещество кислотой или основанием на молекулярном уровне?». Вот уже больше века химики бьются над определением кислот и оснований.
Первая попытка определить понятие кислоты восходит к 1778 году. Антуан Лавуазье смог объяснить, что именно происходит при горении, опровергнув бытовавшую до того теорию о флогистоне. Содержащийся в воздухе газ, который соединяется с веществами, когда они горят, он назвал кислородом — от греческого «производящий кислоту», поскольку он считал (как потом оказалось, ошибочно), что все кислоты содержат кислород.
Определение Аррениуса
Современный подход к этой проблеме впервые сформулировал шведский химик Сванте Аррениус (Svante Arrhenius, 1859–1927). Его определение, выдвинутое в 1877 году, было очень простым: если некоторое вещество при растворении в воде высвобождает ион водорода (то есть протон, Н+), значит это кислота. Если же при растворении в воде высвобождается гидроксид-ион (ОН–), то это основание. Согласно этому определению, аккумуляторная кислота, представляющая собой водный раствор серной кислоты (H2SO4), является кислотой, потому что атомы водорода серной кислоты в растворе становятся ионами водорода. Соответственно, гидроксид натрия (NaOH) является основанием, так как в воде он высвобождает гидроксид-ион. Это определение объясняет, почему кислоты и основания нейтрализуют друг друга. Когда гидроксид-ион встречается с ионом водорода, они соединяются с образованием H2O, обычной воды.
Между прочим, Аррениус активно участвовал в дискуссии о внеземном разуме (см. Парадокс Ферми). Он был сторонником теории панспермии — гипотезы о том, что жизнь с планеты на планету могут переносить микроорганизмы, перемещающиеся через космос, а значит, достаточно было жизни развиться лишь однажды, а не на каждой планете, где она есть. На смену этой гипотезе пришла теория направленной панспермии, в соответствии с которой где-то в Галактике существует цивилизация, которая рассылает зародыши жизни с целью заселения подходящих планет. Однако все эти теории только отодвигают решение проблемы происхождения жизни, потому что всё равно остается вопрос, как жизнь зародилась в самом первом месте.
Парадокс Ферми). Он был сторонником теории панспермии — гипотезы о том, что жизнь с планеты на планету могут переносить микроорганизмы, перемещающиеся через космос, а значит, достаточно было жизни развиться лишь однажды, а не на каждой планете, где она есть. На смену этой гипотезе пришла теория направленной панспермии, в соответствии с которой где-то в Галактике существует цивилизация, которая рассылает зародыши жизни с целью заселения подходящих планет. Однако все эти теории только отодвигают решение проблемы происхождения жизни, потому что всё равно остается вопрос, как жизнь зародилась в самом первом месте.
Определение Брёнстеда—Лаури
Определение Аррениуса довольно точное, но область его применения ограниченна — оно годится только для водных растворов (веществ, растворенных в воде). Вот пример реакции, на которую не распространяется определение Аррениуса: если вы поместите рядом сосуды с соляной кислотой (HCl) и аммиаком (NH3), вы увидите белый дымок над сосудами. Пары аммиака и соляной кислоты смешиваются в воздухе над сосудами, и происходит химическая реакция
Пары аммиака и соляной кислоты смешиваются в воздухе над сосудами, и происходит химическая реакция
NH3 + HCl → NH4Cl,
в которой кислота и основание соединяются с образованием хлорида аммония. Поскольку в этой реакции не участвует вода, определение Аррениуса здесь просто неприменимо.
В 1923 году датский химик Йоханнес Николаус Брёнстед (Johannes Nicolaus Brønsted, 1879–1947) и британский химик Томас Мартин Лаури (Thomas Martin Lowry, 1874–1936) предложили новое определение. В соответствии с ним кислота представляет собой молекулу или ион, способные отдавать протон (то есть ион водорода, H+), а основание представляет собой молекулу или ион, способные принимать протон. Если рассматриваемая реакция протекает в водной среде, это определение по сути то же, что и определение, предложенное Аррениусом, однако оно распространяется также на реакции, протекающие в отсутствие воды, такие как образование хлорида аммония, описанное выше.
Определение Льюиса
Наконец, последнее обобщение сделало определение кислот и оснований не зависящим не только от присутствия воды, но и от образования протонов. Его выдвинул в 1923 году американский химик Гилберт Ньютон Льюис (Gilbert Newton Lewis, 1875–1946). Это определение основано на том, каким способом образуются химические связи в химических реакциях между кислотами и основаниями, а не на том, присоединяются или отдаются протоны. По Льюису, кислота — это химическое соединение, способное принимать электронную пару с последующим образованием ковалентной связи, а основание — это соединение, способное отдавать электронную пару.
Его выдвинул в 1923 году американский химик Гилберт Ньютон Льюис (Gilbert Newton Lewis, 1875–1946). Это определение основано на том, каким способом образуются химические связи в химических реакциях между кислотами и основаниями, а не на том, присоединяются или отдаются протоны. По Льюису, кислота — это химическое соединение, способное принимать электронную пару с последующим образованием ковалентной связи, а основание — это соединение, способное отдавать электронную пару.
Определение Льюиса включает в себя оба более ранних определения, а также объясняет те реакции, в которых не участвует водород. Например, когда диоксид серы реагирует с ионом кислорода с образованием серного ангидрида (эта реакция играет немаловажную роль в образовании кислотных дождей), ион кислорода отдает два электрона для образования ковалентной связи — иными словами, ведет себя как основание, в то время как диоксид серы принимает электроны и, следовательно, ведет себя как кислота. Эта реакция, протекающая без протона и без воды, подходит под определение Льюиса, но не подходит ни под одно из предшествующих определений.
Показатель pH: измерение кислотности
Для водных растворов широко используется система определения концентрации кислоты или основания, которая лучше всего может быть объяснена в терминах теории Брёнстеда—Лаури. В чистой воде в каждый момент времени какие-то молекулы H2O диссоциируют на ионы водорода (H+) и гидроксид-ионы (OH–), и одновременно с этим какие-то соседние ионы H+ и OH– соединяются с образованием молекул воды. Таким образом, в воде всегда присутствуют ионы водорода (протоны). Молярная концентрация (см. Закон Авогадро) водорода в чистой воде составляет 10–7 моль на литр. Это означает, что одна молекула H2O из каждых 10 миллионов находится в форме ионов.
Условились считать, что водородный показатель pH (сокр. от англ. «power of hydrogen» — «степень водорода») чистой воды равен 7 — это математический показатель степени из выражения 10–7, взятый с положительным знаком. Мы можем повысить концентрацию ионов водорода в воде, добавив кислоту. Например, если мы добавим в чистую воду соляную кислоту (HCl), концентрация ионов водорода возрастет. Если мы достигнем точки, в которой молярная концентрация составляет 10–1 моль на литр, мы получим примерное значение кислотности желудочного сока. pH этого раствора составит 1. Таким образом, pH ниже 7 характеризует кислоту, и чем меньше значение pH, тем сильнее кислота.
Мы можем повысить концентрацию ионов водорода в воде, добавив кислоту. Например, если мы добавим в чистую воду соляную кислоту (HCl), концентрация ионов водорода возрастет. Если мы достигнем точки, в которой молярная концентрация составляет 10–1 моль на литр, мы получим примерное значение кислотности желудочного сока. pH этого раствора составит 1. Таким образом, pH ниже 7 характеризует кислоту, и чем меньше значение pH, тем сильнее кислота.
Подобным образом можно понизить концентрацию ионов водорода в чистой воде, добавив основание (ионы OH– основания будут реагировать с ионами H+ с образованием молекул воды). Так, у аммиака, применяемого в домашнем хозяйстве, молярная концентрация ионов водорода составляет всего 10–11 моль на литр, и, следовательно, pH равен 11. А поскольку pH больше 7, это основание.
Общая характеристика кислот — урок. Химия, 8–9 класс.
Кислотами называют сложные вещества, состоящие из атомов водорода, способных замещаться металлами, и кислотных остатков.
Кислотным остатком называют часть молекулы кислоты, соединённую с атомами водорода.
При замещении водорода в кислотах металлами в состав образующихся солей кислотные остатки переходят в неизменном виде. Если кислотный остаток в кислоте соединён с одним атомом водорода, то он одновалентен, если с двумя — двухвалентен, если с тремя — трёхвалентен и т. д.
Валентность кислотного остатка определяется количеством атомов водорода, способных замещаться металлами.
Формулы и названия некоторых кислот приведены в таблице.
Важнейшие неорганические кислоты
Название кислоты | Формула кислоты | Формула кислотного остатка | Название соли этой кислоты |
| Фтороводородная (плавиковая) | HF | −F | Фторид |
| Хлороводородная (соляная) | HCl | −Cl | Хлорид |
| Бромоводородная | HBr | −Br | Бромид |
| Угольная | h3CO3 | =CO3 | Карбонат |
| Кремниевая | =SiO3 | Силикат | |
| Азотная | HNO3 | −NO3 | Нитрат |
Ортофосфорная (фосфорная) | h4PO4 | ≡PO4 | Ортофосфат (фосфат) |
| Серная | h3SO4 | =SO4 | Сульфат |
| Сернистая | h3SO3 | Сульфит | |
| Сероводородная | h3S | =S | Сульфид |
Представителем органических кислот является уксусная кислота Ch4COOH. Хотя в молекуле этой кислоты — четыре атома водорода, только один из них (входящий в состав группы СООН) может быть замещён металлом. Поэтому кислотный остаток уксусной кислоты является одновалентным.
Хотя в молекуле этой кислоты — четыре атома водорода, только один из них (входящий в состав группы СООН) может быть замещён металлом. Поэтому кислотный остаток уксусной кислоты является одновалентным.
Что такое кислотность желудка?
Кислотность желудка — это характеристика концентрации соляной кислоты в желудочном соке. Эту кислоту продуцируют специальные париетальные клетки из желёз желудка. Главные функции соляной кислоты – переваривание белковой пищи, антибактериальное действие, возбуждение работы поджелудочной железы, активация пищеварительных ферментов. А главное, соляная кислота способствует нормальной переработке пищи и ее дальнейшему перемещению в двенадцатиперстную кишку.
Пониженная кислотность, так как и повышенная, абсолютно одинаково причиняют организму чувство дискомфорта в области желудка и приводят к серьезным хроническим заболеваниям органов желудочно-кишечного тракта.
Жидкости в отношении их кислотности считаются: нейтральными при рН =7, кислыми при рН< 7, щелочными при рН> 7.
Максимальная теоретически возможная кислотность в желудке 0,86 рН.
Минимальная теоретически возможная кислотность в желудке 8,3 рН. Нормальная кислотность в просвете тела желудка натощак 1,5–2,0 рН.
Кроме того, в разное время суток и в разных отделах желудка показатели кислотности варьируют.
Причины, ведущие к нарушению кислотности желудка
Увеличивают кислотность:- некачественное питание, нерегулярное потребление еды, злоупотребление кофе, копченостями, диетой;
- существует много болезней, которые требует длительного приема противовоспалительных препаратов;
- стрессовые ситуации;
- курение, избыточное поступление никотина;
- хеликобактерная инфекция.
- хеликобактерная инфекция;
- нарушение пищевого поведения;
- вредные привычки;
- генетические нарушения;
- заболевания печени и поджелудочной железы;
- патология эндокринной системы и обменных процессов.


Как мы видим, причины во многом похожи. Но у каждого конкретного человека они вызовут различные проявления болезни, в зависимости от изначального статуса здоровья человека, от его образа жизни и питания и наличия сопутствующей патологии.
Какие же симптомы помогут заподозрить нарушение кислотности желудка?
Для повышенной кислотности характерно: изжога, боль «под ложечкой», в правом подреберье, характер боли может быть разный (ноющие, приступообразные), отрыжка кислым, жжение в горле и груди, белый налёт на языке. Более поздние симптомы: запоры, кишечная колика.
Для пониженной кислотности характерно: гнилостный запах, отрыжка тухлым яйцом, металлический привкус, поносы и запоры, снижение моторики кишечника, метеоризм, наличие непереваренных остатков пищи в кале. Кроме того, при пониженной кислотности нарушается усвоение витаминов и минералов, переваривание белка. Что в свою очередь проявляется угревой сыпью, ломкостью волос и ногтей, снижением гемоглобина, вялостью, снижением выносливости, частым появлением грибковых и паразитарных заболеваний.
Однако судить только на основании ощущений пациента о наличии нарушения уровня кислотности в желудке судить нельзя. Для постановки правильного диагноза человеку необходимо пройти обследование.
Методы определения кислотности
Для определения кислотности желудочного сока существует ряд методов. Какие-то из них не очень удобны, какие-то не очень точны. В настоящее время используется два основных метода.
Первый из них это определение кислотности непосредственно во время проведения ФГДС. Когда через зонд во время исследования вводится краситель и по изменению цвета судят о кислотности. Но это только косвенное определение. А вот «золотым стандартом», наиболее информативным и физиологический методом является внутрижелудочная рН-метрия. В желудок вводится зонд, что позволяет определить одновременно кислотность в разных зонах желудка.
Из всего вышесказанного можно сделать следующие выводы.
Если Вас беспокоят какие-либо из вышеперечисленных симптомов, то необходимо начать действовать, чтобы сохранить и укрепить своё здоровье.
Вам необходимо обратиться к гастроэнтерологу, который в зависимости от ваших жалоб и симптомов заболевания, назначит необходимое обследование. А по результатам обследования Вы получите конкретные, необходимые именно Вам рекомендации по питанию, изменению образа жизни и получите лекарственные назначения. Ведь недообследованность и самолечение могут привести к непоправимой утрате здоровья. Помните об этом. Ждем Вас в нашей клинике.
Анализ крови на Мочевую кислоту в лаборатории KDL
Мочевая кислота — конечный продукт распада азотсодержащих соединений, называемых пуриновыми основаниями, или пуринами. Эти вещества являются одним из основных компонентов нуклеиновых кислот и входят в состав всех клеток организма. В процессе естественной гибели старых и поврежденных клеток, а также при употреблении определенных продуктов (красное мясо, субпродукты, колбасы и комчености, бобовые, пиво) пурины поступают в печень, где преобразуются в мочевую кислоту, которая высвобождаются в кровь и выводится из организма с мочой.
Повышенный уровень мочевой кислоты в крови (гиперурикемия) может вызвать подагру – заболевание, связанное с нарушением обмена пуриновых оснований. При подагре происходит отложение кристаллов мочевой кислоты в суставах, что сопровождается приступами боли и снижением подвижности вплоть до полной невозможности функционирования сустава. Избыток солей мочевой кислоты может также осаждаться в почках, что приводит к камнеобразованию.
Гиперурикемия может возникать при злоупотреблении богатой пуринами пищей и напитками, при массовой гибели клеток (например, при лечении онкологических заболеваний), при выраженном метаболическом синдроме или под влиянием наследственных факторов. При патологии почек возможна задержка выведения мочевой кислоты из организма.
В каких случаях обычно назначают исследование?
Анализ крови на содержание мочевой кислоты назначается при признаках артрита, в диагностике подагры, а также при следующих состояниях:
- Для контроля уровня мочевой кислоты у людей, проходящих химиотерапию или лучевую терапию при лечении опухолей;
- Для выявления причин образования и рецидивов камней в почках;
- Для мониторинга эффективности лечения подагры.
Что означают результаты теста?
Гиперурикемия развивается в тех случаях, когда образуется избыточное количество мочевой кислоты или почки не справляются с ее выведением. Для определения причин такого состояния необходимы дополнительные исследования.
Пациентам с диагностированной подагрой или наличием уратных (состоящих из мочевой кислоты) камней в почках следует избегать продуктов с высоким содержанием пуринов и алкоголя. Также повышение уровня мочевой кислоты может быть вызвано стрессом, диетами, интенсивными физическими упражнениями и быстрой потерей веса. У некоторых пациентов гиперурикемия может долгое время протекать бессимптомно и выявляться только при сдаче анализа.
Сроки выполнения теста.
Обычно результат можно получить уже на следующий день.
Как подготовиться к анализу?
Следует придерживаться общих правил подготовки к взятию крови из вены. С подробной информацией можно ознакомиться в соответствующем разделе статьи.
Определение pH – simulation, animation – eduMedia
Определение pH – simulation, animation – eduMedia⚠ eduMedia will be under maintenance on June 12, 2021
Краткое содержание
Параметр, характеризующий кислотный, основной (щелочной) или нейтральный характер водного раствора называется pH (водородный показатель).
• pH< 7: кислый раствор
• pH=7 : нейтральный раствор
• pH>7 : основной (щелочной) раствор
Определение pH непосредственно даёт сведения о наличии в растворе некоторых ионов: иона водорода H+ и гидроксид-иона HO—.
- pH< 7:Раствор является кислым и содержит больше ионов водорода H+, чем гидроксид-ионов HO—.
- pH=7 : Нейтральный раствор, в котором столько же ионов водорода H+, сколько и гидроксид-ионов HO—.
- pH>7 : Основной (щелочной) раствор, в котором больше гидроксид-ионов HO— , чем ионов водорода H+.
Водородный показатель (pH) связан с определением концентрации ионов H+. Эти ионы, как и гидроксид-ионы, реагируют со многими веществами, в том числе и с водой. Сильные кислоты или сильные основания (щелочи) являются особо коррозийными. С ними нужно обращаться с большой осторожностью.
Нажать, а затем передвинуть индикаторную бумажку pH для того, чтобы сравнить изменение её окраски.
Цели обучения
- Уметь пользоваться индикаторной бумажкой pH.
- Распределять характерные вещества на шкале pH
- Определить в чём отличие кислого раствора от основного.
- Установить связь между pH и ионами H+ и HO—.
Узнать подробнее
Важной характеристикой водного раствора (на водной основе) является pH. Он характеризует кислую или…
Подпишитесь для доступа к этому разделу!
Поделиться ×
Определение основных органических кислот в различных типах вин после проведения технологических приемов Текст научной статьи по специальности «Прочие сельскохозяйственные науки»
ХИМИЧЕСКИЕ НАУКИ
Ученые записки Крымского федерального университета имени В. И. Вернадского Биология. Химия. Том 3 (69). 2017. № 3. С. 249-256.
УДК 663.22/.253.2:547.477.1:543.544
ОПРЕДЕЛЕНИЕ ОСНОВНЫХ ОРГАНИЧЕСКИХ КИСЛОТ В РАЗЛИЧНЫХ ТИПАХ ВИН ПОСЛЕ ПРОВЕДЕНИЯ ТЕХНОЛОГИЧЕСКИХ ПРИЕМОВ
Аристова Н. И.1, Зайцев Г. П.1, Панов Д. А.2
1ФГБУН «ВННИИВиВ «Магарач» РАН», г. Ялта, Республика Крым, Россия 2Таврическая академия (структурное подразделение) ФГАОУ ВО «Крымский федеральный университет имени В. И. Вернадского», Симферополь, Республика Крым, Россия E-mail: [email protected]
Определена массовая концентрация основных органических кислот в тихих и игристых винах различных производителей методом высокоэффективной жидкостной хроматографии. Рассчитано соотношение винной и яблочной кислот в различных типах вин. Установлено, что массовая концентрация лимонной кислоты в готовой винодельческой продукции после проведения различных технологических приемов не превышает допустимые нормы и соответствует действующей нормативной документации.
Ключевые слова: массовая концентрация, органические кислоты, титруемая кислотность, лимонная кислота, высокоэффективная жидкостная хроматография, регулирование кислотности.
ВВЕДЕНИЕ
Органические кислоты винограда играют большую роль в формировании вкуса и качества вина. Их общее содержание определяют пригодность винограда для получения того или иного типа вина. Недостаточная кислотность делает вкус вина простым и плоским, высокая же приводит к резкому кислому вкусу. Органические кислоты участвуют в создании букета готового вина, придают ему приятную свежесть, защищают вино от бактериальных заболеваний и продлевают срок его хранения. Введение этих кислот в организм человека стимулирует работу поджелудочной железы, способствует лучшему усвоению пищи.
В виноматериалах и винах содержится шесть основных органических кислот: винная, яблочная, молочная, лимонная, уксусная и янтарная. Винная и яблочная кислоты являются основными представителями алифатических кислот виноградных вин, их общая доля составляет 90 % всех кислот в вине. Гармоничное сочетание полноты вкуса и ощущение кислотности обуславливается соотношением между этими кислотами. Если это соотношение (винной кислоты к яблочной) 2:1, то вино будет чрезвычайно кислым. Если повысить долю винной до 3 и выше, то продукт получится высокого качества с гармоничным ароматом и вкусом. Винная кислота,
обладая кислым вкусом, не приводит к появлению посторонних тонов. При высоком же содержании яблочной кислоты вино приобретает тон «зеленой» кислотности. Такие вина требуют дополнительной технологической обработки [1, 2]. В Российской Федерации, согласно правилам производства винодельческой продукции, для повышения кислотности и исправления низкокислотных виноматериалов разрешается добавлять лимонную или винную кислоты не более 2 г/дм3. В России чаще всего практикуется подкисление вина и реже — подкисление сусла [3, 4].
При внесении лимонной кислоты в вино главной целью является не только подкисление, но и образование растворимых комплексов с железом для предотвращения железного касса [1, 3-6]. Кишковский и Мержаниан считают, что устойчивость переокисленных вин к помутнению повышается после подкисления их лимонной кислотой, так как увеличивается агрегативная устойчивость танатов [7]. По концентрации отдельных органических кислот и соотношению между ними можно объективно судить о подлинности виноградных вин.
Для определения органических кислот в винах используют целый ряд физико-химических методов: потенциометрия, хроматография, спектрофотомерия, капиллярный электрофорез и др. В большинстве методов при определении кислот устраняют мешающие компоненты вина или выделяют их с помощью ионного обмена [8, 9-11]. Наиболее эффективным методом при определении слабых кислот в вине является высокоэффективная жидкостная хроматография (ВЭЖХ). Использование этого метода обеспечивает возможность разделения большого количества органических кислот без применения сложной и длительной пробоподготовки. ВЭЖХ позволяет значительно сократить затраты и время проведения анализа [12].
Важно отметить, что в готовой винной продукции регламентируется только лимонная кислота и титруемые кислоты (сумма органических кислот и их кислых солей), поэтому определение лимонной кислоты и основных органически кислот в вине современными методами анализа после проведения технологических приемов, таких как регулирование кислотности, деметаллизация с помощью пищевой лимонной кислоты, является актуальным.
Целью данной работы явилось определение массовой концентрации основных органических кислот в тихих и игристых винах методом ВЭЖХ с использованием хроматографической системы Agilent Technologies (модель 1100) после проведения технологического приема регулирования кислотности с помощью пищевой лимонной кислоты.
МАТЕРИАЛЫ И МЕТОДЫ
В качестве объектов использовали различные типы вин: тихие и игристые, произведенные предприятиями Республики Крым. С целью определения массовых концентраций основных органических кислот были использованы 18 образцов тихих вин и 5 образцов игристых. Отбор проб вин осуществляли по ГОСТ 31730-2012 [13], подготовку проб — по ГОСТ 26671-2014 [14]. Образцы исследуемых игристых вин подвергали процедуре предварительной дегазации на ультразвуковой бане BADELIN SONOREX (Германия). Качественный и количественный состав органических кислот определяли методом высокоэффективной жидкостной с использованием
хроматографической системы Agilent Technologies (модель 1100) с диодно-матричным, рефрактометрическим детекторами аналогично методике [15]. Для разделения органических кислот использовалась карбогидратная хроматографическая колонка «Supelcogel-C610H» размером 7,8*300 мм, заполненная катионообменным полимерным сорбентом зернением 9,0 мкм. Скорость потока элюента — 0,5 см3/мин. и объем пробы 5 мкл. Температура термостата колонки 300 оС, подвижная фаза — 0,1 % раствор ортофосфорной кислоты [10, 11]. Для градуировки прибора использовали стандартные образцы органических кислот квалификации не ниже ч. д. а. Навески образцов растворяли в бидистиллированной воде.
Хроматограммы регистрировали при длине волны 210 нм для органических кислот. Идентификацию компонентов производили по их временам удерживания. Хроматографирование проводили в градиентном режиме. Все определения проводили в трех повторностях. Результаты исследований обрабатывали стандартными методами математической статистики. Относительная погрешность метода составила 2,8-3,0 % при доверительной вероятности Р=0,95.
В ходе проведения технологического приема регулирования кислотности использовали пищевую лимонную кислоту по ГОСТ 31726-2012 [16].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Измерение массовой концентрации основных органических кислот в различных типах вин методом ВЭЖХ проводили в диапазонах от 0,15 до 5 г/дм3. В таблицах 1 и 2 приведены данные по определению содержания основных органических кислот в тихих и игристых винах после проведения технологических приемов.
Так как в готовой винодельческой продукции регламентируется содержание лимонной кислоты (не более 1 г/дм3), то необходимо уделить внимание ее содержанию в исследуемых винах.
Таблица 1
Содержание органических кислот в исследованных тихих винах различных
производств
Массовая концентрация, мг/дм 3 Сумма кислот, мг/дм3
Наименование вина Лимонная Винная Яблочная Молочная + Янтарная Уксусная Отношение гр. 3-4
1 2 3 4 5 6 7 8
Инкерман Мерло столовое 402 2663 554 2477 194 4,8 6290
сухое красное (образец № 1)
Инкерман Алиготе столовое сухое белое
245 3667 745 2760 345 4,9 7762
Продолжение таблицы 1
1 2 3 4 5 6 7 8
Инкерман Совиньон столовое сухое ( образец № 1) 86 2381 456 1979 319 5,2 5221
Инкерман Жемчужина столовое сухое 218 2582 722 2290 298 3,6 6110
Инкерман Вайт полусухое белое 663 2335 1029 1885 218 2,3 6130
Инкерман Шардоне столовое сухое белое 182 3282 783 1509 268 4,2 6024
Инкерман Легенда полусладкое белое 951 2175 808 2482 389 2,7 6805
Инкерман Крымская Ривьера полусладкое белое 982 2391 1673 1741 162 1,4 6949
Инкерман Ркацители сухое столовое 327 3258 1397 738 122 2,3 5842
Инкерман Совиньон+ Рацители столовое сухое 378 2840 1536 850 118 1,8 5722
Инкерман Каберне столовое сухое (образец № 1) 35 3174 415 2853 181 7,6 6658
Инкерман Каберне столовое сухое (образец № 2) 0 2100 417 5272 360 5,0 8149
Инкерман Совиньон столовое сухое ( образец № 2) 415 3584 1371 1557 283 2,6 7210
Инкерман Бастардо столовое сухое красное 29 2700 1089 3188 637 2,5 7643
Евпатория Портвейн 777 белый 614 1140 677 822 175 1,7 3428
Белозерский Херес крепкий 388 1571 2048 2478 333 0,8 6818
Евпатория Портвейн Приморский 380 3790 2730 2840 320 1,4 10060
Гурзуф Кагор ликерное красное 900 1954 1697 590 0 1,2 5141
Таблица 2
Определение массовой концентрации органических кислот в игристых винах
Наименование вина Массовая концентрация, мг/дм3 Отношение гр. 3-4 Сумма кислот, мг/дм3
Лимонная Винная Яблочная Молочная + Янтарная Уксусная
1 2 3 4 5 6 7 8
Новый Свет Пино нуар брют 265 3652 1291 2324 359 2,8 7891
Новый свет брют 269 3582 963 2815 346 3,7 7975
Новый свет экстра брют 300 3301 1088 3022 279 3,0 7990
Новый свет Крымское игристое брют 474 3222 1475 6189 198 2,2 11558
Новосветское полусухое 533 2901 1585 2140 523 1,8 7682
Исходя из табличных данных (Табл. 1), в тихих белых столовых винах значение массовой концентрации лимонной кислоты варьировало от 86 до 982 мг/дм3, в тихих красных столовых — от 0 до 402 мг/дм3, в крепких специальных винах (портвейн, херес) — от 380 до 614 мг/дм3, в ликерных — 900 мг/дм3, а в игристых — от 265 до 533 мг/дм3. Следовательно, значение массовой концентрации лимонной кислоты исследованных образцах тихих и игристых вин находится в пределах допустимой нормы.
На основании проведенных исследований установлены количественные отношения винной кислоты к яблочной. В тихих винах (Табл. 1) это отношение изменяется от 0,8 до 7,7, в игристых винах (Табл. 2, «брют») — от 2,2 до 3,7, а в «Новосветском полусухом» — 1,8.
Одним из основных анализов технохимического контроля состава вин является титруемая кислотность. Из литературных данных известно, что для различных типов вин титруемая кислотность колеблется в определенных пределах: для тихих вин — от 3 до 8 г/дм3, игристых — от 6 до 8,5 г/дм3 [1, 2]. В исследованных образцах тихих вин сумма кислот изменяется в пределах от 3,4 до 8,1 г/дм3, исключение составляет «Портвейн Приморский» (Евпатория) масса кислот — 10,6 г/дм3. Для игристых вин масса кислот около 8 г/дм3, кроме «Крымского игристого» для которого сумма кислот — 11,6 г/дм3.
ЗАКЛЮЧЕНИЕ
Проведенные исследования показали, что содержание лимонной кислоты и титруемая кислотность в различных типах вин, произведенных предприятиями Республики Крым, соответствует действующей нормативной документации: ГОСТ 32030-2013 [17], ГОСТ 52404-2005 [18], ГОСТ 32715-2014 [19], ГОСТ 33336-2015[20].
Список литературы
1. Справочник по виноделию. Изд. 3-е, перераб. и доп./ Под ред. Г. Г. Валуйко, В. Т. Косюры. -Симферополь: Таврида, 2005. — 588 с.
2. Кишковский З. Н. Химия вина / З. Н. Кишковский, И. М. Скурихин. — М.: АгроПромиздат, 1988. -254 с.
3. Сборник основных правил, технологических инструкций и нормативных материалов по производству винодельческой продукции. Разработано Всероссийским научно-исследовательским институтом пивоваренной, безалкогольной и винодельческой промьшленности Россельскохозакадемии, утв. 05.05.98. — М.: Пищепромиздат, 1998. — 242 с.
4. Использование лимонной кислоты для предупреждения железного касса / В. И. Зинченко,
B. Т. Косюра, С. Т. Огородник [и др.] // Садоводство, виноградарство и виноделие Молдавии. -1986. — № 3. — С. 35.
5. Смирнов В. А. Пищевые кислоты / В. А. Смирнов. — М.: Легкая и пищевая промышленность, 1983. — 264 с.
6. Жилякова Т. А. Современные методы контроля показателей качества и безопасности виноградных вин / Жилякова Т. А., Аристова Н. И., Панова Э. П. [и др.] // Ученые записки Таврического национального университета им. В. И. Вернадского. Серия «Биология, химия». — Т. 19 (58). -2006. — № 2. — С. 84-93.
7. Кишковский З. Н. Технология вина / З. Н. Кишковский, А. А. Мержаниан. — М.: Легкая и пищевая промышленность, 1984. — 504 с.
8. Захарова Э. А. Определение общей кислотности и содержания лимонной кислоты в винах потенциометрическим методом / Э. А. Захарова, М. Л. Москалева, Ю. А. Акенеев [и др.] // Журнал аналитической химии. — 2011. — Т. 66, № 9. — С. 964-969.
9. Zoecklein, B. Wine analisis and production / B. Zoecklein, K.C.Fugelsang, B. Gump, F.S. Nury. — N.Y.; Springer US, 1999. — 621 р.
10. Аристова Н. И. Методики выполнения измерений физико-химических показателей для контроля качества винопродукции / Н. И. Аристова // «Магарач»: Виноградарство и виноделие. — 2014. -№ 4. —
C. 36-39. (Aristova N. I. Techniques for the measurement of physical and chemical parameters for the quality control of wine products, «Magarach»: viticulture and winemaking, 4, 36 (2014). (in Russ.)
11. Жилякова Т. А. Определение дополнительных показателей качества и безопасности винодельческой и безалкогольной продукции / Т. А. Жилякова, Н. И. Аристова, Е. В. Дерновая [и др. ] Виноградарство и виноделие : сб.научн. тр. НИВиВ «Магарач». Ялта, 2014. — Том XLIV. -С. 96-99.
12. Захарова А. М. Определение органических кислот, углеводов, подсластителей в пищевых продуктах и биологически активных добавках методом высокоэффективной жидкостной хроматографии / А. М. Захарова, Л. А. Карпова, И. Л. Гринштейн // Аналитика и контроль. -2013. — Т. 17, № 2. — С. 204-210.
13. ГОСТ 31730-2012 Продукция винодельческая. Правила приемки и методы отбора проб. — М.: Стандартинформ, 2013. — 12 с.
14. ГОСТ 26671-2014 Продукты переработки фруктов и овощей, консервы мясные и мясорастительные. Подготовка проб для лабораторных анализов. — М.: Стандартинформ, 2014. — 7 с.
15. Р 4.1. 1672-03 Руководство по методам контроля качества и безопасности биологически активных добавок к пище. — М.: Федеральный центр Госсанэпидемнадзора Минздрава России, 2004. — 184 с.
16. ГОСТ 31726-2012 Добавки пищевые. Кислота лимонная безводная Е 330.Технические условия -М.: Стандартинформ, 2014. — 19 с.
17. ГОСТ 32030-2013 Вина столовые и виноматериалы столовые. Общие технические условия. — М.: Стандартинформ, 2014. — 7 с.
18. ГОСТ 52404-2005 Вина специальные и виноматериалы специальные. Общие технические условия. — М.: Стандартинформ, 206. — 8 с.
19. ГОСТ 32715-2014 Вина ликерные. Вина ликерные защищенных географических указаний, вина ликерные защищенных наименований места происхождения. Общие технические условия — М.: Стандартинформ, 2014. — 6 с.
20. ГОСТ 33336-2015. Вина игристые. Общие технические условия. — М.: Стандартинформ, 2015. — 11 с.
DETERMINATION OF MAJOR ORGANIC ACIDS IN DIFFERENT TYPES OF WINES AFTER CARRYING OUT TECHNOLOGICAL METHODS
Aristova N.I.1, Zaytsev G.P.1, Panov D.A.2
1Government-Financed Establishment of the Republic of the Crimea «National Research Institute for
Vine and Wine «Magarach», Crimea, Russian Federation
2V. I. Vernadsky Crimean Federal University, Simferopol, Crimea, Russian Federation
E-mail: [email protected]
Wine materials and wines contains six major organic acids — tartaric, malic, lactic, citric, acetic and succinic. Tartaric and malic acids are the main representatives of aliphatic acids of wines, their combined share is 90% of all acids in wine. In the Russian Federation according to the rules of wine production to increase the acidity and correcting low acid wine you can add citric or tartaric acid not more than 2 g/dm3.
For the determination of organic acids in wines may using a number of physico-chemical methods: potentiometric, chromatography, spectrophotometry, capillary electrophoresis, etc. The most effective method in the determination of weak acids in wine is high performance liquid chromatography (HPLC).
The aim of this work was the determination of the mass concentration of main organic acids in still and sparkling wines by HPLC using a chromatographic system of Agilent Technologies (model 1100), after the technical admission control acidity with food citric acid.
The objects used were different types of wines: still and sparkling, produced by enterprises of the Republic of Crimea. To determine the mass concentrations of the major organic acids were used 18 samples of wine and 5 samples of sparkling.
Chromatograms were recorded at a wavelength of 210 nm for organic acids. The identification of components was carried out according to their retention time. Chromatography was performed in gradient mode. All definitions were carried out in three replicates. The research results were processed by standard methods of mathematical statistics. The relative error of the method was 2.8-3% at a confidence probability P=0,95.
Based on the data in the quiet white table wines value of the mass concentration of citric acid ranged from 86 to 982 mg/dm3, in a quiet red table — from 0 to 402 mg/dm3, special strong wines (port, sherry) — from 380 to 614 mg/dm3, liquor — 900 mg/dm3, and sparkling from 265 to 533 mg/dm3.
In the quiet wines the ratio of tartaric acid to malic is changed from 0.8 to 7.7, in the sparkling wines from 2.2 to 3.7, and «Novyi Svet, semi-dry» of 1.8. Studies have shown that the content of citric acid and titratable acidity in different types of wines produced by enterprises of the Republic of Crimea corresponds to the current normative documents GOST 32030-2013, GOST 52404-2005, GOST 32715-2014, GOST 33336-2015.
Keywords: mass concentration, organic acids, titratable acidity, citric acid, highperformance liquid chromatography.
References
1. Valuyki G. G., Koury V. T. Handbook of winemaking. Ed. 3-e, Rev., 588 p. (Tavrida, Simferopol, 2005). (in Russ.)
2. Kishkovsky Z. N., Skurikhin I. M. Wine chemistry, 254 p. (Agropromizdat, Moscow, 1988). (in Russ.)
3. A collection of basic rules, technological instructions and regulations in force for the production of wine products. Developed all-Russia scientifically-research Institute brewing, nonalcoholic and wine promyshlennosti of Rosselkhozakademii, approved. 05.05.98, 242 p. (Piwepromizdat, Moscow, 1998). (in Russ.)
4. Zinchenko, V. I., Kosura, V. T., Ogorodnik, S. T., Kochetkov, T. P., Krechetova, V. V. The use of citric acid to prevent the iron ticket office, Horticulture, viticulture and winemaking Moldova, 3, 35 (1986). (in Russ.)
5. Smirnov V. A. Food acids, 264 p. (Light and food industry, Moscow, 1983). (in Russ.)
6. Zhilyakova T. A. Modern methods of monitoring of indicators of quality and safety of grape wine, Scientific Notes of Taurida National V. I. Vernadsky University — Series: Biology, Chemistry, 19 (58) 2, 84 (2006). (in Russ.)
7. Kishkovsky Z.N., Merzhanian A.A. Technology of wine, 504 p. (Light and Food Industry, Moscow, 1984). (in Russ.)
8. E. A. Zakharova, M. L. Moskaleva, J. A. Akeneev, E. S. Moiseeva, G. B. Slepchenko, N. P. Picula Determination of total acidity and the content of citric acid in wines by potentiometric method. Journal of analytical chemistry, 66 (9), 964 (2011). (in Russ.)
9. Zoecklein, B. Wine analisis and production / B. Zoecklein, K.C.Fugelsang, B. Gump, F.S. Nury. — N.Y.; Springer US, 1999. — 621 р.
10. Aristova N.I. Techniques for the measurement of physical and chemical parameters for the quality control of wine products, «Magarach»: viticulture and winemaking, 4, 36 (2014). (in Russ.)
11. Zhilyakova T. A., Aristova N. And. Sod E., Alder J. L., Guseva I. P., Zaitsev, G. P. The definition of additional indicators of quality and safety of wine and soft drinks, «Magarach»: viticulture and winemaking, ХЫУ, 96 (2014). (in Russ.)
12. A. M. Zakharov, L. A. Karpova, I. L. Greenstein Determination of organic acids, carbohydrate sweeteners in foods and dietary supplements by high-performance liquid chromatography analysis and control, 17, 2, 204 (2013). (in Russ.)
13. GOST 31730-2012 wine Production. Acceptance rules and sampling methods, 12 p. (Standartinform, Moscow, 2013). (in Russ.)
14. GOST 26671-2014 Products of processing fruits and vegetables, canned meat and meat plant. Preparation of samples for laboratory analyses, 7 p. (Standartinform, Moscow, 2014). (in Russ.)
15. P 4.1. 1672-03 Quality control methods Manual and safety of biologically active additives to food, 184 p. (Federal Center gossanepidemnadzora Russian Ministry of Health, Moscow, 2004). (in Russ.)
16. GOST 31726-2012food Supplements. Anhydrous citric acid E330. Specifications, 19 p. (Standartinform, Moscow, 2014). (in Russ.).
17. GOST 32030-2013 Wine canteen and wine canteen. General technical conditions, 7 p. (Standartinform, Moscow, 2014). (in Russ.),
18. GOST 52404-2005 Wine special, and wine special. General technical conditions, 8 p. (Standartinform, Moscow, 2006). (in Russ.)
19. GOST 32715-2014 Wine liqueur. Liqueur wines of protected geographical indications, protected liqueur wines of the appellations of origin. General specifications, 6 p. (Standartinform, Moscow, 2014). (in Russ.)
20. GOST 33336-2015. The wine sparkling. General technical conditions, 11 p. (Standartinform, Moscow, 2015). (in Russ.)
5 ключевых факторов, влияющих на кислотность в органической химии
Пять ключевых факторов, влияющих на кислотность
Сегодня мы поговорим о том, что стоит за тенденциями кислотности для различных молекул, и обсудим наиболее важные факторы, определяющие эти значения.
Раньше я писал школьным тоном о том, что pKa является одним из самых важных показателей, которым вы можете научиться в органической химии, и незнание некоторых основных значений pKa перед экзаменом во многом похоже на то, чтобы подойти к покерному столу, не зная значения рук: вы потеряете рубашку.
Давайте быстро рассмотрим основы кислотности и основности. Вот сокращенная версия:
- Кислоты Бренстеда являются донорами протонов, кислоты Льюиса — акцепторами электронных пар . Обратное: основание Бренстеда = акцептор протона, основание Льюиса = донор электронной пары.
- Основание конъюгата — это то, что вы получаете при удалении протона (H +) из соединения. Например, HO — — сопряженная основа воды. O 2- является сопряженным основанием HO —.И наоборот, конъюгированные кислоты — это то, что вы получаете, когда добавляете протон к соединению. Конъюгированная кислота воды H 3 O + .
- Быстрая проверка: кислотный или щелочной pH 1? pKa похож на pH в том смысле, что низкие (и даже отрицательные значения) обозначают сильные кислоты. Это потому, что pKa основывается на равновесии:
- Согласно этому все, что стабилизирует конъюгированное основание, увеличивает кислотность. Следовательно, pKa также является мерой того, насколько стабильно сопряженное основание.Другими словами, сильные кислоты имеют слабые сопряженные основания, и наоборот.
Разобравшись с этим, приступим.
Содержание
- Фактор 1 — начисление.
- Фактор № 2 — Роль атома
- Фактор № 3 — Резонанс
- Фактор № 4 — Индуктивные эффекты
- Фактор № 5 — Орбитали
1. Фактор № 1 — Заряд.
Удаление протона H + снижает формальный заряд атома или молекулы на одну единицу.Это, конечно, легче всего сделать, когда атом изначально несет заряд +1, и становится все труднее, когда общий заряд становится отрицательным. Тенденции кислотности отражают это:
Обратите внимание, что как только конъюгат основания (B-) становится отрицательным, второе депротонирование приводит к образованию дианиона (B 2-). Хотя это далеко не невозможно, формирование дианиона может быть затруднено из-за накопления отрицательного заряда и соответствующего электронного отталкивания, которое возникает в результате.
2.Фактор №2 — роль атомаЭтот момент вызывает большую путаницу из-за наличия двух, казалось бы, противоречащих друг другу тенденций.
Вот первый момент: кислотность увеличивается по мере прохождения по строке в периодической таблице. В этом есть смысл, правда? Имеет смысл, что HF более электроотрицателен, чем h3O, Nh4 и Ch5, из-за большей электроотрицательности фтора по сравнению с кислородом, азотом и углеродом. Фтор, несущий отрицательный заряд, — это счастливый фтор.
Но вот, казалось бы, странная вещь.Сам по себе HF не является «сильной» кислотой, по крайней мере, не в том смысле, что он полностью ионизируется в воде. HF — более слабая кислота, чем HCl, HBr и HI. Что тут происходит?
Вы можете привести два аргумента в пользу этого. Первая причина связана с более короткой (и более прочной) связью H-F по сравнению с более крупными галогенидами водорода. Второй связан со стабильностью конъюгированного основания. Фторид-анион F (-) — крошечный и злобный зверь с наименьшим ионным радиусом среди всех других ионов, несущих единственный отрицательный заряд.Следовательно, его заряд распределен по меньшему объему, чем у более крупных галогенидов, что энергетически невыгодно: во-первых, F (-) требует сольватации, что приведет к более низкому члену энтропии в Δ G.
Обратите внимание, что эта тенденция также сохраняется для h3O и h3S, причем h3S примерно в 10 миллионов раз более кислый.
3. Фактор №3 — РезонансОгромным стабилизирующим фактором для сопряженной базы является то, что отрицательный заряд может быть делокализован посредством резонанса.Классические примеры — фенол (C6H5OH), который примерно в миллион раз более кислый, чем вода, и уксусная кислота (pKa ~ 5).
Однако будьте осторожны — для π-системы недостаточно просто находиться рядом с протоном — электроны сопряженного основания должны находиться на орбитали, которая обеспечивает эффективное перекрытие (для подлого вопроса с подвохом в этом Вены, которые обычно блокируют выпуски Гарварда, смотрите здесь.)
4. Фактор № 4 — Индуктивные эффектыЭлектроотрицательные атомы могут притягивать к себе отрицательный заряд, что может привести к значительной стабилизации сопряженных оснований.Посмотрите эти примеры:
Как и ожидалось, этот эффект будет связан с двумя основными факторами: 1) электроотрицательностью элемента (чем больше электроотрицательный, тем более кислотный) и расстоянием между электроотрицательным элементом и отрицательным зарядом. .
5. Фактор № 5 — ОрбиталиОпять же, кислотность хорошо связана со стабильностью конъюгированного основания. И стабильность сопряженного основания зависит от того, насколько хорошо оно может приспособиться к своей новообретенной паре электронов.В эффекте, похожем на электроотрицательность, чем больше s-характер на орбитали, тем ближе электроны будут к ядру и тем ниже их энергия (= стабильная!).
Посмотрите разницу между pKa ацетилена и алканов — 25! Это 10 в степени 25, как «в 100 раз больше числа Авогадро». Просто чтобы дать вам представление о масштабе. Это удивительная вещь в химии — диапазон и силы различных явлений внушает трепет.
На самом деле я нашел мнемонику, которая может помочь вам запомнить эти эффекты. Это приписывается доктору Кристине Пруис, старшему преподавателю Темпе Университета штата Аризона.
C harge
A tom
R esonance
D ipole I nduction
O rbitals
= .
Будьте осторожны с мнемоникой, но готово.
Основность — обзор | Темы ScienceDirect
6.2.4 Как?
Основность данного углеродного материала в принципе будет определяться количеством и прочностью основных участков, присутствующих на его поверхности. Понимание основности углерода оказалось далеко не простой задачей. Для этого есть три основные причины: (i) основные центры на углероде сосуществуют с кислотными центрами, которые могут действовать как источники протонов, т.е. углеродные поверхности являются амфотерными; (ii) внутренняя сложность, химически говоря, углеродных поверхностей, которая делает довольно труднодостижимой задачей прямую идентификацию возможных основных сайтов; и (iii) трудности в различении кислот, которые физически адсорбируются только на углеродных поверхностях, и кислот, которые химически нейтрализуются углеродными поверхностями.Таким образом, большинство экспериментальных исследований основности углерода ограничиваются грубым описанием общих кислотно-основных взаимодействий между углеродным материалом и жидким или газообразным источником протонов. Поскольку адсорбция касается взаимодействия адсорбатов с твердыми поверхностями, экспериментальные методики, разработанные для проверки основности углерода, можно рассматривать как исследования адсорбции на углеродных материалах с использованием конкретного зонда (источника протонов). Это описание позволяет нам подчеркнуть важный вопрос во время оценки основности углерода с помощью текущих экспериментальных процедур: основность углерода не только зависит, как указано выше, от количества и силы основных участков, но также в значительной степени зависит от доступности этих участков.В этом смысле все связанные с диффузией особенности, которые упускаются из виду при работе с однородными растворами, могут фактически определять результат измерений основности углерода. Это особенно, но не исключительно важно при характеристике основности пористых материалов.
На сегодняшний день наиболее распространенные методики проверки основности углерода включают источники протонов в водных растворах, от относительно простых определений pH до потенциометрического титрования [5–8]. Все эти методики адаптированы из традиционных методов титрования, разработанных для гомогенных водных растворов.Поскольку углеродные материалы в принципе не растворяются в воде, эти гомогенные растворы заменяются твердыми суспензиями, что приводит к ряду последствий, которые необходимо учитывать. Во-первых, как только что упоминалось, параметры, связанные с диффузией, такие как размер твердых частиц, количество тестируемого твердого вещества на объем водного раствора, температура, ионная сила и размер ионов влияют на этот тип измерений. Во-вторых, для понимания основности углерода требуется знание взаимодействия воды и углерода. Это взаимодействие, которое описывается в терминах гидрофильности или гидрофобности, также должно играть решающую роль при измерении основности углерода в водных суспензиях.Например, можно задаться вопросом, подходит ли методология на водной основе для тестирования основных гидрофобных участков (т.е. базальных плоскостей чистого углерода) на углеродных поверхностях [9].
Измерения основности углерода, основанные на адсорбции газов или паров в качестве источников протонов, встречаются реже. Это важно, поскольку способность углеродной поверхности действовать как поглотитель протонов может быть различной в (сухой) газовой фазе и жидкой фазе. Объемный или гравиметрический анализ адсорбции кислых газов или паров является подходящим методом для изучения основности углерода, хотя исследования, похоже, ограничиваются высокопористыми материалами [10,11].Другие подходы, основанные на адсорбции различных газовых или паровых зондов, альтернативно использовались для получения информации об основности углерода, включая твердые вещества с низкой удельной площадью. Было продемонстрировано, что газо-твердотельная хроматография (или, как недавно было переименовано, обратная газовая хроматография), является мощным инструментом для мониторинга изменений основности (по Льюису) различных углеродных материалов [12–15]. Чувствительность метода позволяет работать при очень низкой концентрации адсорбата, практически в режиме закона Генри.После принятия донорно-акцепторных концепций для описания энергетики взаимодействия газа и твердого тела элюирование выбранных молекул с хорошо известными физико-химическими свойствами может в конечном итоге привести к параметризации основности углеродной поверхности (донорного числа).
Адсорбционная энергия, в газовой или жидкой фазе, также использовалась для учета основности углерода. Калориметрические методы позволяют обнаруживать изменения энергии взаимодействия (т. Е. Теплоты адсорбции) между кислотным зондом и поверхностью углерода [16–18].Если предполагается, что текстурные свойства ряда материалов очень похожи, то эти изменения могут быть связаны с основным характером твердой поверхности.
Адсорбция выбранных зондов также может отслеживаться различными спектроскопическими методами. Подход относительно прост и основан либо на обнаружении молекулярных свойств зонда, адсорбированного на поверхности углерода, либо на изменениях свойств этих поверхностей после адсорбции. FTIR, твердотельный ЯМР, XPS и ESR — вот некоторые примеры полезных методов для этой цели.В принципе, должна быть возможность количественно определить количество основных сайтов на углероде. Однако это довольно оптимистичный сценарий для большинства углеродных материалов из-за как матричных эффектов, так и сложности такой матрицы (то есть углеродного материала). Полуколичественный анализ основных сайтов может быть в конечном итоге выполнен после разработки очень специфических поверхностных реакций, в которых только основные сайты или даже определенный тип основного сайта склонны реагировать с выбранной молекулой [19,20]. Обычно в таких случаях требуется предварительное принятие моделей для описания основных участков на углероде (см. Ниже).
Вернувшись в водные растворы, основные угли, действующие как поглотители протонов, будут демонстрировать избыток положительного заряда на своей поверхности. Измерение этих зарядов позволит определить основность материалов. Обычные методики включают оценку электрокинетического потенциала (ζ-потенциала) и массовое титрование [21]. Сравнение результатов, полученных с помощью этих двух методов измерения поверхностного заряда, т. Е. Так называемой изоэлектрической точки и точки нулевого заряда, соответственно, представляет собой хороший пример актуальности местоположения основных участков при испытании углеродных материалов.Таким образом, для пористого углерода электрокинетические измерения, в отличие от массового титрования, в значительной степени определяются основными группами на внешней поверхности материала. Другими словами, основные участки, расположенные на внутренних порах, не «доступны» при зондировании с помощью этого метода. Как следствие, электрокинетические измерения и массовое титрование одного и того же (пористого) углерода могут предоставить информацию об основности поверхности, которая может качественно отличаться (кислотный или щелочной характер) [21,22].
Электрохимические измерения также могут быть инструментом для определения основных характеристик углеродного материала, хотя требуются дополнительные знания, чтобы установить прямую взаимосвязь между электрохимическим откликом углерода и их основным характером [23,24]. Когда порошковый электрод из активированного угля погружается в первый раз в раствор электролита, он проявляет большую электрохимическую емкость, около 100 Ф · г -1 и более, из-за большой поверхности пор и адсорбции ионов электролита.Такая емкость двойного слоя примерно соответствует 1 ммоль / г -1 адсорбированных ионных частиц, что позволяет предположить, что значительная часть кислотно-основных частиц в водном растворе могла бы взаимодействовать с поверхностными группами углерода, создавая двойной электрический слой, а не чем реакция протонного обмена с этими группами. Другими словами, кислотно-основная буферная емкость некоторых углеродных материалов, вероятно, связана с их электрохимической емкостью, и, следовательно, как эффекты физической адсорбции, так и собственная основность углеродных функциональных групп будут играть важную роль в электрохимическом отклике углеродного электрода. .
Основность — обзор | ScienceDirect Topics
Отношение бикарбоната к кремнезему Гаррелса-Маккензи имеет значение не только для связи состава силикатных пород в континентальной литосфере с составом подземных вод. Среднее время пребывания грунтовых вод составляет 2 × 10 4 год, что является достаточным временем для того, чтобы грунтовые воды уравновесились с надземной атмосферой. В отсутствие углекислого газа pH грунтовых вод будет в диапазоне от 10 до 11 (см. Пример 6.2), что намного выше, чем было обнаружено при анализе грунтовых вод.Влияние растворенного диоксида углерода будет подробно рассмотрено в Разделе 6.4; на данный момент достаточно признать, что растворение диоксида углерода (вставка 6.2) и реакция гидролиза (6.R12), по-видимому, достигают равновесия в течение среднего времени пребывания или, по крайней мере, близко приближаются к равновесию (вставка 6.4).
Пример 6.2Пример Постоянная ссылка
http://soilenvirochem.net/ofc13K
Рассчитайте pH раствора 1,0 × 10 -3 M Na 2 CO 3 (водный), используя RICE табличный метод и следующие реакции гидролиза оснований и коэффициент равновесной реакции.
CO32− (водный) + h3O (l) ↔HCO3− (водный) + OH− (водный) Kb1 = 2,14 × 10−4 = aHCO3 − ⋅aOH − aCO32−
Шаг 1. Приготовьте RICE таблица (Таблица 6.7), представляющая гидролиз Na 2 CO 3 (водн.) в растворе.Карбонат-анионы CO 3 2- (водн.) Подвергаются двух стадиям гидролиза основания. Вторая константа основного гидролиза намного меньше, чем первая константа основного гидролиза, поэтому концентрация гидроксид-иона OH — (водн.) Почти полностью возникает на первой стадии гидролиза.
Таблица 6.7. RICE Таблица для гидролиза карбонатной основы
| Реакция → | CO 3 2- (водн.) (Моль-дм -3 ) | H 2 O (л) | ↔ | HCO 3 — (водный) (моль дм −3 ) | OH — (водный раствор) (моль дм −3 ) |
|---|---|---|---|---|---|
| Исходный | 10 −4 | 0 | 0 | ||
| Реакция | — x | + x | + x | ||
| 6 — | 900 Равновесие | x | x |
Шаг 2. Используя термины из таблицы RICE , напишите выражение равновесного отношения для первой стадии гидролиза основания и решите полином второго порядка, используя квадратное уравнение.
Шаг 3. Смоделируйте pH раствора 1,0 × 10 −3 M Na 2 CO 3 (водный), используя бикарбонат HCO 3 – в качестве карбонатного компонента.Минималистическая симуляция без учета баланса зарядов и ионов-наблюдателей содержит два компонента: HCO3– и H +.Установка начальной концентрации HCO 3 — равной 1,0 × 10 −3 M обеспечивает правильный общий карбонат, но только половину щелочности 1,0 × 10 −3 M Na 2 CO 3 (водн.) раствор. Добавление дополнительной щелочности в моделирование ChemEQL требует замены компонента H + на OH−, установка режима (OH−) = total и присвоение ему начальной концентрации 1,0 × 10 −3 M. Общая щелочность с этими компонентами и исходной желаемая концентрация 1.0 × 10 −3 Концентрация М.
Смоделированное значение pH составляет 10,51, по сравнению с оценкой pH 10,57 в таблице RICE .
Вставка 6.4Простая модель Гиббса
Хотя химическое выветривание (раздел 6.3.1), несомненно, является основным фактором, определяющим состав грунтовых вод, поверхностные воды подвергаются другому значительному процессу: испарению. Совместное воздействие выветривания и испарения горных пород было наглядно проиллюстрировано на двух графиках «бумеранга», которые впервые появились в статье Гиббса (1970).
Гиббс (1970) использовал единицы концентрации ppm (мг кг -1 ) для всех растворенных веществ и нанес на ординату обоих графиков общее количество растворенных твердых веществ (TDS) (единицы: -1 мг кг). 8 Гиббс построил соотношение массовых долей f Na, Ca на абсциссе первого графика и соотношение массовых долей f Cl, HCO 3 на абсциссе второго графика. Данные, представленные на этих графиках, были взяты из основной подборки составов поверхностных вод (Ливингстон, 1963) и исследования 16 притоков реки Амазонки (Гиббс, 1972).
fNa, Ca≡w (Na) w (Na) + w (Ca) fCl, HCO3≡w (Cl) w (Cl) + w (HCO3)
Геологическая служба США (USGS) Professional Paper 440-G (Livingstone , 1963) перечисляет 412 анализов, подходящих для построения графиков, но Гиббс решил нанести на график анализы «135 крупных озер и рек по всему миру» (Гиббс, 1992), а не результаты всех анализов. График всего набора данных Ливингстона (1963) не обнаруживает следа «бумеранга», столь очевидного в работах Гиббса (Гиббс, 1970, 1971, 1992). Несмотря на неоднократную защиту модели (Гиббс, 1971, 1992), более сложная картина вырисовывается из всеобъемлющего анализа глобального состава поверхностных вод и обоснованных возражений против модели (Фет и Гиббс, 1971; Килхэм, 1990; Эйлерс и др., 1992).Гиббс (1970) определил три конечные точки на обоих графиках «бумеранга»: воды с преобладанием метеоритов, воды с преобладанием выветривания и воды с преобладанием испарения / химического осаждения.
Конечная точка крайне низкой солености возникает там, где отношения массовых долей f Na, Ca и f Cl, HCO 3 приближаются к единице. В поверхностных водах в этой конечной точке преобладают метеорные воды. Основными ионами в водах с чрезвычайно низкой соленостью, в которых преобладают метеориты, являются хлоридные соли, характерные для морских аэрозолей.
Вторая конечная точка возникает при «умеренной» солености — 100–300 ppm TDS — где отношения массовых долей f Na, Ca и f Cl, HCO 3 приближаются к нулю. В этой конечной точке химическое выветривание пород преобладает над составом поверхностных вод. Основные ионы в водах с преобладанием выветривания средней солености образуются в результате реакций выветривания (6.R11) и (6.R14).
Третья конечная точка приходится на чрезвычайно высокую соленость, характерную для основных океанов и соленых озер, где отношения массовых долей f Na, Ca и f Cl, HCO 3 отклоняются в сторону единицы. .Комбинированное воздействие испарения и карбонатных осадков доминирует над составом поверхностных вод в этой конечной точке. Основными ионами в водах с испарением с чрезвычайно высокой соленостью / с преобладанием химических осадков являются хлоридные соли, характерные для морской воды.
График всех 412 анализов Ливингстона (1963) показывает треугольник с вершинами в трех конечных точках, определенных Гиббсом (1970). Процесс, отсутствующий в простой модели Гиббса, заключался в эрозии эвапоритовых образований подземными водами (Ван Денбург и Фет, 1965; Фет и Гиббс, 1971).В таблице 6.8 перечислены EC на четырех участках USGS вдоль реки Пекос в Нью-Мексико. Соленость реки Пекос удваивается с 1,86 дСм −1 в Санта-Роза, штат Нью-Мексико, до 3,53 дСм м −1 на станции Темный каньон около Карлсбада, штат Нью-Мексико — расстояние до реки примерно 450 км. Это увеличение солености могло быть результатом испарения, но модель Гиббса не может объяснить двукратное увеличение солености между Карлсбадом и Малагой — расстояние до реки всего 43 км — до 7,14 дСм м -1 .Соленость реки Пекос снова увеличивается вдвое до 15,04 дСм м −1 в Ред-Блафф, штат Нью-Мексико, на расстоянии 34 км от реки.
Таблица 6.8. Средняя электрическая проводимость (dS · м −1 ) проб воды, собранных на четырех участках отбора проб поверхностных вод USGS вдоль реки Пекос, Нью-Мексико, США
| Станция | Идентификационный номер станции | Река-километр (км ) | Электропроводность (dS m −1 ) | |||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Санта-Роуз | 08383000 | 1190 | 1.860 | |||||||||||||||||||||||||||||||||||||||||||||||
| Карлсбад | 08405200 | 739 | 3.525 | |||||||||||||||||||||||||||||||||||||||||||||||
| Малага | 08406500 | 696 | 7,139 | Красный Карбонатные осадки, вызванные выветриванием и испарением, как первоначально определил Гиббс (1970), оказывают сильное влияние на поверхностные и поровые воды почвы кислотно-основной химический состав .Сброс соленых грунтовых вод в реку Пекос (Ван Денбург и Фет, 1965; Фет и Гиббс, 1971) и другие поверхностные воды — не говоря уже о просачивании грунтовых вод с поверхности суши — может иметь сильное влияние на соленость в определенных ландшафтах выше и за пределами солености увеличивается из-за испарения.Кислоты и основанияСогласно теории Бренстеда, кислота является донором протонов , а основание является акцептором протонов . В кислотно-основной реакции каждая сторона равновесия имеет кислотный и основной реагент или продукт, и они могут быть нейтральными частицами или ионами. Структурно связанные пары кислота-основание, такие как {H-A и A: (-) } или {B: (-) и B-H}, называются парами конъюгатов . Вещества, которые могут служить как кислотами, так и основаниями, например вода, называются амфотерными . Кислотность Поскольку эти исследования обычно экстраполируются на высокие разведения, молярная концентрация воды (55,5) постоянна и может быть исключена из знаменателя. Полученное значение K называется константой кислотности , K a . Очевидно, что сильные кислоты имеют больше K и , чем более слабые кислоты.Действительно, если HA полностью ионизируется в растворе, K a стремится к бесконечности ([HA] почти равно нулю). Из-за очень большого диапазона концентраций кислоты (более 10 40 ) обычно используется логарифмическая шкала кислотности (pK a ). Более сильные кислоты имеют меньшие или более отрицательные значения pK и , чем более слабые кислоты. Во всех приведенных выше примерах вода действует как общее основание, причем самая слабая кислота и самое слабое основание будут преобладать и обязательно будут находиться на одной и той же стороне равновесия).В идеале следует использовать концентрацию кислоты не более 0,1 М. Поскольку вода является амфотерной, она может подвергаться автопротолизу, как показано в следующем уравнении. Автоионизация показана как Keq, и если рассматривать концентрацию воды, как и раньше, константа кислотности (K a ) для воды обозначается K w (водная постоянная). Таким образом, pH чистой воды составляет 7,0. Диапазон выделяемых сильных кислот ограничен. Таким образом, кислоты, которые сильнее, чем катион гидроксония, H 3 O (+) , и слабые кислоты, имеющие сопряженные основания сильнее, чем гидроксид-анион, OH (-) , не могут быть измерены непосредственно в водном растворе.Если сильная кислота (HA) имеет Ka = 100, 0,1 М раствор в воде будет полностью ионизирован, давая концентрацию гидроксония 100 и концентрацию [HA] 10 -4 . Аналогичный раствор кислоты с концентрацией в 10 3 раз неопределяемым образом отличался бы из-за выравнивающего эффекта растворителя. Термин выравнивающий эффект относится к влиянию амфотерного растворителя, такого как вода, на измерения кислотности и основности. Сила сильной кислоты выравнивается основностью растворителя, а основность сильных оснований также ограничивается кислотностью растворителя. Поскольку многие органические реакции протекают либо в водной среде (живые клетки), либо гасятся или обрабатываются в воде, важно учитывать, как кислотно-щелочная равновесная смесь конъюгата изменяется с изменением pH. Эту информацию предоставляет простая связь, известная как уравнение Хендерсона-Хассельбаха . Когда pH водного раствора или смеси равен pK a кислотного компонента, концентрации кислотных и основных конъюгированных форм должны быть равны (логарифм 1 равен 0).Если pH снижен на две или более единицы по сравнению с pK a , концентрация кислоты будет выше 99%. С другой стороны, если pH (относительно pK a ) повышается на две или более единицы, концентрация основания конъюгата будет более 99%. Следовательно, смеси кислых и некислотных соединений легко разделяются путем регулирования pH водного компонента при двухфазной экстракции растворителем. Основность Большинство простых алкиламинов имеют pK a в диапазоне от 9,5 до 11,0, а их водные растворы являются основными (имеют pH От 11 до 12, в зависимости от концентрации). Первые четыре соединения в следующей таблице, включая аммиак, попадают в эту категорию. Последние шесть соединений (цветные ячейки) являются значительно более слабыми основаниями в результате трех факторов.Первый из них — гибридизация азота. В пиридине азот sp 2 гибридизован, а в нитрилах (последняя запись) sp-гибридный азот является частью тройной связи. В каждом из этих соединений (заштриховано красным) несвязывающая электронная пара локализована на атоме азота, но увеличение s-символа приближает его к ядру азота, уменьшая его тенденцию связываться с протоном. Во-вторых, анилин и п-нитроанилин (первые две структуры, закрашенные зеленым) являются более слабыми основаниями из-за делокализации несвязывающей пары электронов азота в ароматическое кольцо (и нитрозаместитель).Это та же делокализация, которая приводит к активации бензольного кольца в сторону электрофильного замещения. В самом деле, анилин является более слабым основанием, чем циклогексиламин, примерно в миллион раз, благодаря тому же фактору фенол является более сильной кислотой, чем циклогексанол. Эта делокализация электронной пары сопровождается определенной степенью регибридизации атома азота аминогруппы, но делокализация электронной пары, вероятно, является основным фактором пониженной основности этих соединений. Подобная делокализация электронных пар ответственна за очень низкую основность (и нуклеофильную реакционную способность) амидных атомов азота (последняя структура, закрашенная зеленым). Теория ЛьюисаСогласно теории Льюиса, кислота является акцептором электронной пары , а основание является донором электронной пары . Базы Льюиса также являются базами Бронстеда; однако многие кислоты Льюиса, такие как BF 3 , AlCl 3 и Mg 2+ , не являются кислотами Бренстеда. Продукт кислотно-основной реакции Льюиса представляет собой нейтральный, диполярный или заряженный комплекс, который может быть стабильной ковалентной молекулой. Как показано в верхней части следующего рисунка, координационное ковалентное связывание фосфорного основания Льюиса с борной кислотой Льюиса создает комплекс, в котором формальный заряд бора отрицательный, а заряд фосфора положительный.В этом комплексе бор приобретает конфигурацию неоновой валентной оболочки, а фосфор — конфигурацию аргона. Два примера кислотно-основного равновесия Льюиса, которые играют роль в химических реакциях, показаны в уравнениях 1 и 2 ниже. В первом примере электронодефицитный атом алюминия связывается с ковалентным атомом хлора, разделяя одну из его несвязывающих валентных электронных пар, и, таким образом, достигает октета аргоноподобной валентной оболочки.Поскольку это разделение является односторонним (хлор вносит вклад в оба электрона), как показано, и алюминий, и хлор имеют формальные заряды. Если связь углерод-хлор в этом комплексе разрывается, и оба связывающих электрона остаются с более электроотрицательным атомом (хлор), углерод принимает положительный заряд. Мы называем такие виды углерода как карбокатионы . Карбокатионы также являются кислотами Льюиса, как показывает обратная реакция. Если заместители (R) на этих атомах не велики, комплекс будет находиться в равновесии.Однако стерические затруднения объемных заместителей могут препятствовать образованию комплексов. Полученная смесь несвязанных пар кислота / основание Льюиса была названа «фрустрированной» и демонстрирует необычные химические и каталитические свойства. Комбинация P (трет-Бутил) 3 и B (C 6 F 5 ) 3 является примером. Терминология, относящаяся к номенклатуре кислот и оснований Льюиса, часто используется химиками-органиками. Здесь термин электрофил соответствует кислоте Льюиса, а нуклеофил соответствует основанию Льюиса. Электрофил : электронодефицитный атом, ион или молекула, которые имеют сродство к электронной паре и будут связываться с основанием или нуклеофилом. Некоторая путаница в различении основности (основной силы) и нуклеофильности (нуклеофильной силы) неизбежна. Поскольку основность — это менее хлопотное понятие; с него удобно начинать. Основность относится к способности основания принимать протон . Основность может быть связана с pK a соответствующей конъюгированной кислоты.Самые сильные основания имеют самые слабые сопряженные кислоты и наоборот . В кислотно-щелочном равновесии самая слабая кислота и самое слабое основание будут преобладать, и они обязательно будут на одной стороне равновесия. Растворители, такие как уксусная кислота, ацетонитрил и нитрометан, часто используются для изучения очень сильных кислот. Аналогичным образом, очень слабокислые растворители, такие как диметилсульфоксид (ДМСО), ацетонитрил, толуол, амины и аммиак, могут быть использованы для изучения кислотности очень слабых кислот.Измерения в этих растворителях можно экстраполировать на воду, и многие из них имеют большую погрешность. Как для кислот, так и для щелочей указанные значения pK и , экстраполированные на воду, являются приблизительными. Таблица некоторых основных реагентов приведена ниже. Диапазон основностей замечателен, он охватывает более пятидесяти степеней десяти!
Нуклеофильность — более сложное свойство. Обычно это относится к скорости реакции замещения в галогенсодержащий атом углерода эталонного галогенида алкила, такого как CH 3 -Br. Таким образом, нуклеофильность реагента Nu: (-) в следующей реакции замещения изменяется, как показано на диаграмме ниже. Диапазон реакционной способности этих реагентов превышает 5000 раз, причем тиолат является наиболее реактивным.Ясно, что есть существенные различия между этими нуклеофильностями и основностями, обсужденными выше.
Для двух или более молекул, содержащих нуклеофильные атомы одного вида и заряда, более сильное основание обычно является более сильным нуклеофилом.Таким образом, 2,2,2-трифторэтоксид (pK a 12) является более слабым основанием и нуклеофилом, чем этоксид (pK a 16). Заметное исключение из этого правила возникает, когда вицинальный (соседний) атом несет несвязывающую электронную пару. Двумя распространенными примерами этого исключения, называемого альфа-эффект , являются гидроксид-ион (pK a 15,7) по сравнению с ионом гидропероксида (pK a 11,6) и аммиак (pK a 9,3) по сравнению с гидразином (pK a 8,0). В каждой из этих пар более слабое основание — более сильный нуклеофил. Сольватация нуклеофильных анионов заметно влияет на их реакционную способность. Приведенные выше нуклеофильности были получены в результате реакций в растворе метанола. Полярные протонные растворители, такие как вода и спирты, сольватируют анионы за счет взаимодействия водородных связей, как показано на диаграмме справа. Эти сольватированные частицы более стабильны и менее реакционны, чем несольватированные «голые» анионы. Полярные апротонные растворители, такие как ДМСО (диметилсульфоксид), ДМФ (диметилформамид) и ацетонитрил, не сольватируют анионы почти так же хорошо, как метанол, но обеспечивают хорошую сольватацию сопутствующих катионов.Следовательно, большинство обсуждаемых здесь нуклеофилов быстрее реагируют в растворах, приготовленных из этих растворителей. Такие эффекты растворителя более выражены для небольших основных анионов, чем для крупных слабоосновных анионов. Таким образом, для реакции в растворе ДМСО наблюдается следующий порядок реактивности:
В отличие от порядка в метаноле (показанном выше), этот порядок примерно соответствует порядку увеличения основности.Нейтральные основания, такие как аммиак и простые первичные и вторичные амины, имеют сходную основность (pKa = от 9 до 11) и также являются хорошими нуклеофилами. В реакциях с бромистым метилом в метаноле их нуклеофильность больше, чем Cl (-) , и меньше, чем N 3 (-) . В слабоосновных апротонных растворителях они реагируют с умеренными электрофилами, такими как алкилгалогениды (Cl, Br и I) и карбонильные группы (альдегиды, кетоны, сложные эфиры и ангидриды), давая четвертичные соли из первых и имины, енамины и амиды из последних. .Третичные амины и объемные вторичные амины являются более бедными нуклеофилами и ведут себя исключительно как основания. Эффекты растворителя Кислоты
Есть несколько факторов, которые влияют на измерения кислотности, сделанные в воде и других растворителях. Эти: 1. Ионизация кислоты дает ионы. Растворители с большими диэлектрическими постоянными способствуют образованию и разделению заряда.2. Конъюгированная кислота из растворителя представляет собой катион, который стабилизируется за счет ассоциации с другими молекулами растворителя, явление, называемое сольватацией. 3. Основание, сопряженное с кислотой, представляет собой анион, который в большинстве случаев также стабилизируется сольватацией. В качестве примеров рассмотрим следующие данные, полученные для шести сильных кислот в трех распространенных растворителях.
Все три растворителя имеют довольно высокие диэлектрические постоянные, но вода (ε = 80) значительно больше, чем ДМСО (ε = 46,7) или ацетонитрил (ε = 37,5). Более высокая кислотность (более низкий pK a ) последних четырех соединений в воде может быть отнесена на счет диэлектрической разницы; тем не менее, лучшая сольватация анионов, обеспечиваемая водой, считается основным фактором.И ДМСО, и ацетонитрил являются плохими растворителями для сольватации анионов, поэтому показанное выше ионизационное равновесие будет смещено влево. Полезно сначала сосредоточиться на различиях pK a между водой и ДМСО. Мы делаем это, потому что вода и ДМСО имеют схожую основность, поэтому стабилизация видов кислоты, сопряженной с растворителем, не должна существенно отличаться. HBr и HCl отображают pK a s в ДМСО, которые примерно на 9-10 раз больше, чем в воде, а более слабая кислота HF второго периода показывает еще большее изменение (pK a = 3.17 в воде и 15 в ДМСО). Все сопряженные основания этих кислот представляют собой одноатомные анионы. Поскольку отрицательный заряд локализован, стабилизация анионов путем сольватации, вероятно, является наиболее важным фактором в установлении положения этих равновесий. Напротив, сопряженные основания из серной кислоты и трифторуксусной кислоты внутренне стабилизированы делокализацией заряда по двум или трем атомам кислорода. Следовательно, важность сольватации менее важна, и изменение кислотности в ДМСО снижается до 4-х кратных десяти.
Большинство органических соединений являются гораздо более слабыми кислотами, чем сильные кислоты, рассмотренные выше. Поэтому представляет интерес определить изменения кислотности, которые имеют место, когда такие соединения исследуются в тех же трех растворителях. Первые четыре соединения в этой таблице (заштрихованные светло-зеленым) представляют собой слабые кислоты с сопряженными основаниями, в которых заряд локализован на одном атоме (O, C и S). Замена воды в растворителе на ДМСО снижает кислотность на 10–12 десятичных степеней для атомов второго периода (C и O) и на 6.5 для третьего периода сульфгидриловой кислоты. Так же, как и в более кислых случаях, рассмотренных выше, сольватация сопряженных основных анионов является важным стабилизирующим фактором в воде, хотя более низкая плотность заряда серы снижает ее значимость. Дальнейшее увеличение pK a , которое происходит в растворах ацетонитрила, относительно ДМСО, довольно равномерное (от 10 до 12 единиц) и немного больше, чем сдвиг, обнаруженный для сильных кислот. За исключением производного ацетилена, все предыдущие соединения являются гетероатомными кислотами.Подобные измерения для группы активированных угольных кислот показывают согласие с предыдущим анализом и иллюстрируют способ, которым делокализация анионного заряда снижает pK — разницу между измерениями воды и ДМСО. Обратите внимание на близкую идентичность второго и третьего примеров в следующей таблице. Как и ожидалось, увеличение pK на при переходе в ацетонитрил из ДМСО остается примерно постоянным (около 11).
Основания
Ранее определенные факторы, которые влияли на кислотность, теперь могут быть повторно исследованы для этого нового равновесия. 1. Ионы присутствуют в обеих частях уравнения. Следовательно, различия в диэлектрической проницаемости растворителей должны быть относительно несущественным фактором. В следующей таблице приведены значения pK и для некоторых важных азотистых оснований, некоторые структуры которых показаны справа.В примерах триэтиламина и DABCO в верхней части таблицы катионный заряд относительно локализован на одном атоме азота. Заряд в лутидине может быть делокализован на кольцевых атомах углерода за счет ароматической стабилизации. Последние три примера представляют собой соединения, в которых заряд делокализован за счет резонанса (протонирование происходит у голубого азота) или внутренней водородной связи (протонная губка).
Указанное выше влияние растворителя на кислотность и основность может вызвать неожиданные изменения в простых кислотно-щелочных равновесиях. Если уксусная кислота и триэтиламин смешиваются вместе в воде, происходит быстрый перенос протона с почти количественным образованием ацетата триэтиламмония. Конъюгированные кислоты различаются по силе на 10 6 , и самая слабая из них находится в равновесии.Когда те же два соединения объединяются в растворе ДМСО, перенос протонов незначителен, поскольку уксусная кислота теперь более чем в 10 3 раз слабее кислоты, чем катион триэтиламмония. Функция кислотности ХамметаИзмерения ионизации водных растворов кислот (и оснований) обычно ограничиваются диапазоном pH от 1,5 до 13. Относительные значения более сильных кислот (и оснований) требуют использования других растворителей, а из-за различий в диэлектрической проницаемости сольватация, и прочие отклонения от идеала, устойчивые отношения не достигаются.Выход из этой ситуации был предложен Луи Хэмметом (Колумбийский университет). Хэмметт предложил использовать серию слабых индикаторных оснований, в которых соотношение [B] / [BH + ] можно было определить колориметрически из-за отчетливо окрашенного конъюгата кислоты. Когда отношение [B] к [BH + ] становится особенно малым и экспериментально невозможно точно измерить, другой, менее щелочной индикатор, перекрывающий первый, может служить для расширения диапазона кислотности. Дополнительные более слабые индикаторные основания позволили бы тогда перепрыгнуть через шкалу кислотности, увеличив ее примерно в 10 10 раз.Для этого определяли функцию кислотности H o , как показано с помощью следующего уравнения. В идеале H o обеспечивает количественную меру силы кислоты для неводных и концентрированных водных кислот Бренстеда. Функция кислотности — это мера кислотности среды или системы растворителей, обычно выражаемая в терминах ее способности отдавать протоны основанию с известной основностью. Преобладающая разновидность кислоты в разбавленных водных растворах — H 3 O + , поэтому шкала pH на сегодняшний день является наиболее часто используемой функцией кислотности в таких системах.В концентрированной серной кислоте преобладающим видом кислоты является H 3 SO 4 + , гораздо более сильная кислота, таким образом, H o является расширением шкалы pKa для очень сильных кислот. Ключевым фактором для измерения значимого H o является выбор подходящего растворителя. Идеально. желателен слабоосновный протонный растворитель, имеющий большую диэлектрическую проницаемость, значительную степень автопротолиза и хорошую сольватацию ионов. Нетрадиционным выбором Хамметта была безводная серная кислота .Несмотря на очевидные опасения по поводу коррозионной активности, гигроскопичности и вязкости, этот выбор оказался вдохновляющим, о чем свидетельствуют следующие характеристики. Поскольку следы воды нежелательны, необходимо титрование SO 3 (дымящая серная кислота) до максимальной температуры плавления.
В следующих таблицах перечислены некоторые индикаторные основания Hammett и H o некоторых очень сильных кислот или «суперкислот». Значение pKa индикаторных оснований определяли в водных растворах сильных кислот H 2 SO 4 и HClO 4 . Значения Ho были определены в безводной серной кислоте, и значения выше -16 являются оценочными.
Прочные основанияХимикам-органикам нужны основы разной силы в качестве реагентов, адаптированных к требованиям конкретных реакций.Нейтральные соединения азота могут охватывать диапазон основности примерно 10 10 , как показано в следующей таблице. Пиридин обычно используется в качестве акцептора кислоты в реакциях с образованием побочных продуктов минеральных кислот. Его основность и нуклеофильность могут быть изменены стерическими препятствиями, как в случае лутидина (pK a = 6,3), или резонансной стабилизацией, как в случае 4-диметиламинопиридина (pK a = 9,2). Основание Хюнига относительно ненуклеофильно (из-за стерических затруднений), и, как DBU, часто используется в качестве основания в реакциях элиминирования E2, проводимых в неполярных растворителях.Основание Бартона представляет собой сильное, слабонуклеофильное нейтральное основание, которое используется в случаях, когда электрофильное замещение DBU или других аминовых оснований является проблемой. Самыми сильными основаниями, доступными химикам-органикам, являются соли алкоксидов щелочных металлов, такие как трет-бутоксид калия (pK a = от 16 до 19), алкилы щелочных металлов, такие как реактивы Гриньяра, н-бутиллитий и димсил натрия (pK a ). = От 28 до 40+), а также амидные соли щелочных металлов, такие как LDA (pK a = от 30 до 40) и гидрид натрия (pK a ~ 42).Все эти мощные основания являются потенциальными нуклеофилами (некоторые в большей степени, чем другие) и имеют частично ионные связи с металлами. Основания в этих диапазонах pKa используются для превращения слабых кислот C-H в пригодные карбанионные металлические реагенты.
Неионные сверхоснования Кинетическая кислотностьНаиболее распространенная кислотно-основная терминология, pK a , отражает равновесную кислотность, экстраполированную или нормированную на воду.В следующем уравнении основание B: (-) M (+) отрывает протон от кислоты HA с образованием конъюгированной пары кислота-основание (A: (-) M (+ ) и BH). Скорость прямого отрыва протона составляет k f , а скорость обратного переноса протона составляет k r . Этот вид равновесия обычно характеризуется константой равновесия K eq , которая представляет собой отношение констант скорости (k f / k r ).Если H-A является более слабой кислотой, чем H-B, равновесие будет находиться слева, и K экв. будет меньше 1.
В случаях, когда H-A намного слабее, чем H-B, K eq может быть слишком маленьким для измерения, но при определенных обстоятельствах можно определить скорость прямого отрыва протона.Если меченый изотопом конъюгат кислоты основания используется в качестве растворителя для реакции (B-D в следующих уравнениях), то любое происходящее отщепление протона будет отмечено преобразованием H-A в D-A. Уравнение, закрашенное зеленым сверху, показывает начальную потерю протона, а второе уравнение описывает быстрое дейтерирование промежуточного сопряженного основания, A: (-) . По мере протекания этих реакций реагент H-A будет все больше обозначаться как D-A, и скорость изотопного обмена будет указывать на кинетическую кислотность H-A .Предполагается, что кинетическая кислотность примерно пропорциональна равновесной (термодинамической) кислотности, но это не всегда верно.
Следующая диаграмма представляет собой поучительный пример этих принципов.Первое уравнение, выделенное желтым цветом, дает важную информацию о тяжелой воде (оксид дейтерия), которая будет использоваться в качестве растворителя в нашем эксперименте. Тяжелая вода во многих отношениях похожа на воду, но она на 10% плотнее и в десять раз слабее кислоты. 1 молярная концентрация дейтероксида натрия будет служить основанием, а эквимолярное количество 3,3-диметил-1-бутина будет служить слабой кислотой. Самый кислый водород в этом углеводороде (окрашен в красный цвет) находится в C-1. На практике нам необходимо использовать сорастворитель для полного растворения углеводорода в тяжелой воде, но это было опущено, чтобы упростить обсуждение. Зеленая заштрихованная рамка содержит уравнения, которые помогают нам интерпретировать экспериментальные результаты. Чтобы оценить равновесную кислотность субстрата, нам необходимо измерить константу равновесия K eq для начального кислотно-основного равновесия, показанного в верхней части заштрихованной рамки. Поскольку мы знаем K a 3,3-диметил-1-бутина и тяжелой воды, мы можем оценить K eq , разделив первое (10 -25 ) на второе (10 -17 ).Этот расчет показывает K eq , который было бы трудно измерить напрямую из-за его небольшой величины (10 -8 ). В самом деле, равновесная концентрация ацетилид-аниона оценивается всего в 2 * 10 -10 M. Этот пример также демонстрирует пределы подхода изотопного обмена. Подложка из 3,3-диметил-1-бутина также имеет девять других атомов водорода (окрашенных в оранжевый цвет), которые не обмениваются на дейтерий в этих условиях. Мы знаем, что эти водороды гораздо менее кислые (K a ок.10 -48 ), и интересно рассмотреть их возможное участие в кислотно-основных реакциях с помощью предыдущего анализа. Расчетное значение K экв. для такого образования карбаниона составляет примерно 10 -30 с учетом девятикратного увеличения концентрации. Это означает концентрацию одного карбаниона на каждые 10 9 литр раствора. Кинетический анализ также обескураживает. Константа форвардной ставки оценивается в 10 -20 Ms -1 .Следовательно, время, необходимое для обмена половины этих атомов водорода на дейтерий, составит около 100 веков! Путем сравнения скоростей водородного обмена для различных соединений в идентичных условиях можно составить таблицы относительных кинетических кислотностей.Об интересном примере такого исследования сообщалось для группы нитроалканов, имеющих кислые α-атомы водорода. Удаление α-водорода основанием приводит к образованию конъюгированного основания, называемого аци-анионом , как показано здесь.
Исследования изотопного обмена, катализируемого основаниями, в соединениях, включающих более одного набора кислых водородов, дают дополнительную информацию о создании и использовании нуклеофильных конъюгированных оснований. Кетоны представляют собой множество примеров образования региоизомерного енолятного основания, и на следующей диаграмме показаны два таких случая. Как отмечалось в исследовании нитроалканов, атомы водорода на α-метильной группе обмениваются быстрее, чем на более замещенных α-атомах углерода.Уравнения на диаграмме показывают только исходный продукт от единственного обмена. Эти продукты содержат дополнительные α-водороды, которые также обмениваются в последующих реакциях такого рода, так что полная замена всех α-водородов дейтерием происходит за короткое время. При повторном нажатии кнопки Toggle Equations выше будут отображаться относительные скорости обмена α-водорода для некоторых замещенных циклогексанонов. И снова менее замещенные α-углеродные атомы обмениваются быстрее, но обнаружено, что более высокозамещенные еноляты преобладают в условиях равновесия.Третье нажатие кнопки Toggle Equations отобразит энергетический профиль для случая 2-метилциклогексанона, который должен прояснить различие между кинетической и равновесной кислотностью. Также показаны два других примера. Эти дисплеи можно периодически переключать. Большинство углеродных кислот образуют сопряженные основания, которые стабилизируются делокализацией заряда на соседних гетероатомах. Эта резонансная стабилизация требует значительной структурной реорганизации исходного соединения, что, в свою очередь, создает энергетический барьер, замедляющий скорость отрыва протонов.Например, альфа-углерод кетона или сложного эфира должен подвергаться повторной гибридизации при образовании енолят-аниона. Стереоэлектронные требования этого изменения были описаны выше, и неудивительно, что образование енолят-аниона происходит намного медленнее, чем перенос эквивалентных протонов между спиртами и другими гидроксильными соединениями. Скорости депротонирования фенола и нитрометана, соединений с почти идентичными pK a (10,0), являются поучительным примером этого фактора структурной реорганизации.Кислый протон в феноле связан с кислородом, поэтому депротонирование требует небольшого изменения структуры и происходит очень быстро. Нитрометан — это угольная кислота. Депротонирование до аци-аниона включает значительные структурные изменения и в миллион раз медленнее, чем образование фенолята. Эти структурные изменения показаны на следующей диаграмме. Обратите внимание, что электронная пара O-H в феноле остается в основном на кислороде в соответствующем сопряженном основании, тогда как электронная пара C-H в нитрометане преимущественно смещена на кислород в его сопряженном основании (окрашен в синий цвет).Обозначенные здесь тенденции немного упрощены, поскольку не учитывались влияния растворителей и катионов. Эта страница является собственностью Уильяма Ройша. Комментарии, вопросы и ошибки следует присылать по адресу: Факторы, определяющие кислотную силуВо-первых, мы сосредоточимся на отдельных атомах и подумаем о тенденциях, связанных с положением элемента в периодической таблице. Мы будем использовать в качестве наших первых моделей простые органические соединения этан, метиламин и метанол, но эти концепции в равной степени применимы и к более сложным биомолекулам, таким как боковые цепи аланина, лизина и серина. Мы можем видеть четкую тенденцию кислотности, когда мы движемся слева направо по второй строке периодической таблицы от углерода к азоту и кислороду. Ключом к пониманию этой тенденции является рассмотрение гипотетического конъюгированного основания в каждом случае : чем более стабильно (слабее) конъюгированное основание, тем сильнее кислота . Посмотрите, где заканчивается отрицательный заряд в каждой сопряженной базе. В этил-анионе отрицательный заряд несет углерод, в то время как в анионе метиламина и анионе метоксида заряды расположены на азоте и кислороде соответственно.Вспомните периодическую тенденцию электроотрицательности (раздел 2.3A): она также увеличивается по мере того, как мы движемся слева направо по строке, что означает, что кислород является наиболее электроотрицательным из трех, а углерод — наименее. Чем более электроотрицателен атом, тем лучше он способен нести отрицательный заряд. . Таким образом, метоксид-анион является наиболее стабильным (с наименьшей энергией, наименее основным) из трех сопряженных оснований, а этил-анион является наименее стабильным (с наибольшей энергией, наиболее основным). Мы можем использовать тот же набор идей, чтобы объяснить разницу в основности между водой и аммиаком. Изучая значения pK и для соответствующих конъюгированных кислот, мы знаем, что аммиак более щелочной, чем вода. Кислород, как более электроотрицательный элемент, более плотно связан со своей неподеленной парой, чем азот. Таким образом, неподеленная пара азота с большей вероятностью разорвется и образует новую связь с протоном — другими словами, она более проста. Еще раз, более реакционноспособное (более сильное) основание конъюгата означает менее реакционноспособное (более слабое) конъюгированное основание. При движении по вертикали в пределах данного столбца периодической таблицы мы снова наблюдаем четкую периодическую тенденцию изменения кислотности.Лучше всего это иллюстрируется галогенидами: основность, как и электроотрицательность, увеличивается по мере продвижения вверх по столбцу. И наоборот, кислотность в галогенкислотах увеличивается по мере продвижения вниз на колонки. Чтобы понять эту тенденцию, мы еще раз рассмотрим устойчивость сопряженных оснований. Поскольку фтор является наиболее электроотрицательным элементом галогена, можно ожидать, что фторид также будет наименее основным ионом галогена. Но на самом деле это наименее стабильный, и самый базовый! Оказывается, что при вертикальном движении в периодической таблице размер атома превосходит его электроотрицательность в отношении основности.Атомный радиус йода примерно в два раза больше, чем у фтора, поэтому в ионе йода отрицательный заряд распространяется на значительно больший объем: Это иллюстрирует фундаментальную концепцию органической химии, которая достаточно важна, чтобы выделить ее красным: Электростатические заряды, положительные или отрицательные, более стабильны, когда они «растянуты», чем когда они ограничены одним атомом . Мы увидим, как эта идея выражается снова и снова в ходе нашего исследования органической реакционной способности во многих различных контекстах.Пока это понятие применяется только к влиянию радиуса атома на стабильность аниона. Поскольку фторид является наименее стабильным (наиболее основным) из конъюгированных оснований с галогенидами, HF является наименее кислой из галогенокислот, лишь немного сильнее, чем уксусная кислота. HI, с pK и около -9, является одной из самых сильных известных кислот. Что еще более важно для изучения биологической органической химии, эта тенденция говорит нам о том, что тиолы более кислые, чем спирты. Например, pK a тиоловой группы в боковой цепи цистеина составляет приблизительно 8.3, в то время как pK a для гидроксила в боковой цепи серина составляет порядка 17. Еще раз повторю: сила кислоты увеличивается по мере того, как мы перемещаемся вправо по строке периодической таблицы и по мере того, как мы движемся вниз по столбцу. ПримерИзобразите структуру основания конъюгата, которое образовалось бы, если бы указанное ниже соединение реагировало с 1 молярным эквивалентом гидроксида натрия: Решение Эффект резонансаВ предыдущем разделе мы сосредоточили наше внимание на периодических тенденциях — различиях в кислотности и основности между группами, в которых обменный протон был связан с разными элементами.Теперь пришло время подумать о том, как структура различных органических групп влияет на их относительную кислотность или основность, даже когда мы говорим об одном и том же элементе, действующем как донор / акцептор протонов. Первая модельная пара, которую мы рассмотрим, — это этанол и уксусная кислота, но сделанные нами выводы будут одинаково справедливы для всех групп спирта и карбоновых кислот. Несмотря на то, что обе являются кислородными кислотами, значения pK a этанола и уксусной кислоты сильно различаются.Что делает карбоновую кислоту намного более кислой, чем спирт? Как и раньше, начнем с рассмотрения сопряженных оснований. У обоих видов отрицательный заряд конъюгированного основания удерживается кислородом, поэтому периодические тенденции не могут быть вызваны. Однако для уксусной кислоты есть ключевое различие: можно нарисовать резонансный вклад, в котором отрицательный заряд локализован на втором кислороде группы. Две резонансные формы для сопряженного основания равны по энергии в соответствии с нашими «правилами резонанса» (раздел 2.2С). Как вы можете вспомнить, это означает, что отрицательный заряд ацетат-иона не находится ни на одном, ни на другом кислороде: скорее, он распределяется между ними двумя. Химики используют термин «делокализация заряда» для описания этой ситуации. В этоксид-ионе, напротив, отрицательный заряд «заблокирован» на единственном кислороде — ему больше некуда деваться. А теперь пора вспомнить утверждение из предыдущего раздела, которое было настолько важным, что оно было напечатано жирным шрифтом в отдельном абзаце — на самом деле, это настолько важно, что мы просто скажем это еще раз: «Электростатические заряды. , будь то положительные или отрицательные, более стабильны, когда они «разбросаны», чем когда они ограничены одним атомом.«Теперь мы видим эту концепцию в другом контексте, где заряд« распространяется »(другими словами, делокализуется) за счет резонанса , а не просто за счет размера задействованного атома. Делокализация заряда за счет резонанса оказывает очень сильное влияние на реакционную способность органических молекул, достаточное для того, чтобы учесть разницу более чем в 12 pK a единиц между этанолом и уксусной кислотой (и помните, pK a — это логарифмическое выражение , так что мы говорим о разнице между константами кислотности двух молекул более чем на 10 12 ).Ацетат-ион намного более стабилен, чем этоксид-ион, все из-за эффектов резонансной делокализации. Эффект резонанса также хорошо объясняет, почему атом азота является основным, когда он находится в амине, но , а не основным, когда он является частью амидной группы. Напомним, что в амиде существует значительный характер двойной связи углерод-азотной связи из-за второго резонансного фактора, в котором неподеленная пара азота является частью p-связи. В то время как неподеленная электронная пара аминного азота «застревает» в одном месте, неподеленная пара на амидном азоте делокализована за счет резонанса.Обратите внимание, что в этом случае мы расширяем наше центральное утверждение, чтобы сказать, что плотность электронов — в форме неподеленной пары — стабилизируется за счет резонансной делокализации, даже если здесь нет отрицательного заряда. Вот еще один способ подумать об этом: неподеленная пара на амидном азоте недоступна для связывания с протоном — эти два электрона слишком «удобны», будучи частью делокализованной системы пи-связывания. Напротив, неподеленная пара на аминном азоте не является частью делокализованной p-системы и очень готова к образованию связи с любым кислотным протоном, который может находиться поблизости. Часто требуется тщательное размышление, чтобы предсказать наиболее кислый протон в молекуле. Аскорбиновая кислота, также известная как витамин С, имеет pK — , равное 4,1. В этой молекуле четыре гидроксильные группы — какая из них наиболее кислая? Если мы рассмотрим все четыре возможных сопряженных основания, мы обнаружим, что есть только одно, для которого мы можем делокализовать отрицательный заряд по двум атомам кислорода. ПримерРасположите перечисленные ниже соединения от наиболее кислых до наименее кислых и объясните свои рассуждения. Решение Сравните значения pK a для уксусной кислоты и ее моно-, ди- и трихлорированных производных: Присутствие хлоринов явно увеличивает кислотность группы карбоновой кислоты, но аргумент здесь не имеет отношения к резонансной делокализации, потому что для хлорированных молекул нельзя выделить дополнительные резонансные составляющие. Скорее, объяснение этого явления связано с так называемым индуктивным эффектом .Атом хлора более электроотрицателен, чем водород, и, таким образом, способен «индуцировать» или «притягивать» электронную плотность к себе, от карбоксилатной группы. Фактически, атомы хлора способствуют дальнейшему расширению электронной плотности сопряженного основания, что, как мы знаем, имеет стабилизирующий эффект. В этом контексте заместитель хлора называется электроноакцепторной группой . Обратите внимание, что эффект понижения pK a каждого атома хлора, хотя и значительный, не столь драматичен, как эффект делокализационного резонанса, проиллюстрированный разницей в значениях pK a между спиртом и карбоновой кислотой.В общем, резонансные эффекты более мощные, чем индуктивные эффекты . Индуктивный электроноакцепторный эффект хлора осуществляется за счет ковалентных связей, и его влияние заметно уменьшается с расстоянием — таким образом, хлор на два атома углерода от группы карбоновой кислоты оказывает меньшее влияние по сравнению с хлором, находящимся на расстоянии одного атома углерода. ПримерПример Оцените соединения ниже от наиболее кислых до наименее кислых и объясните свои рассуждения. Решение АвторыROCO Кислотная основа: Самый простойМетодический подход работает лучше всего, но вы можете попробовать два разных подхода. Метод 1 — самое сильное основание имеет самый слабый конъюгат кислота Сначала просканируйте молекулу на предмет всех негалогенных атомов с неподеленными парами (обычно N и O).Во-вторых, представьте, что протонируете каждый атом-кандидат и вытягиваете его сопряженную кислоту. В-третьих, определите самую слабую конъюгированную кислоту. Протонированный атом в самой слабой сопряженной кислоте является самым основным атомом в исходной молекуле. Пример поможет вам разобраться в следующих шагах: При сканировании молекулы на атомы с неподеленными парами получаются два кандидата: кольцо N и O. Затем мы рисуем конъюгированную кислоту каждой группы.(Я нарисовал только кислотную функциональную группу, чтобы сэкономить место.) Наконец, мы оцениваем относительную кислотность этих воображаемых конъюгированных кислот. Катион пиридиния или pyrH + (pKa +5) является гораздо более слабой кислотой, чем ROh3 + (pKa -2), поэтому N является более основным атомом. Когда исходная молекула вступает в реакцию с кислотой, протоны будут избирательно переходить на N, потому что они будут менее способны покинуть этот атом. Этот метод может показаться немного запутанным, но после небольшой практики он станет довольно простым. Метод 2 — сканирование молекулы на предмет наиболее сильной известной основной группы Современные химики-органики склонны сосредотачиваться на силе кислоты. Когда нам нужно подумать о прочности основания, мы вспоминаем старую поговорку: «, конъюгированная кислота сильного основания является слабой, а — наоборот, ». Вот почему я впервые показал вам, как найти самый основной атом, глядя на различные сопряженные кислоты. Однако вы также можете научиться распознавать основные группы и узнать их относительную реактивность.Если у вас есть эта информация под рукой, вы можете сканировать молекулу непосредственно на наличие этих групп. Обращаясь к молекуле выше, я знаю, что пиридин (C6H5N) является более сильным основанием, чем вода (которую я считаю разновидностью спирта), поэтому N является самым основным атомом в этой молекуле. Если вы не можете вспомнить свойства общих базовых групп, можно также прибегнуть к некоторым общим принципам: # 1 Важность — отрицательно заряженные основания сильнее нейтральных.Положительно заряженные молекулы редко бывают основными. # 2 Важность — ищите деактивирующие группы, включая RSO2, RC = O и Ph. Эти группы деактивируют базу. # 3 Важность — при прочих равных основание N сильнее, чем основание O. # 4 Важность — в категории функциональной группы используйте эффекты заместителя для сравнения оснований. Каждый из этих принципов сопровождается важными ограничениями.Они объясняются в эссе «Определите наиболее кислотный H молекулы», и вам следует обратиться к нему за подробным объяснением. Когда вы читаете это эссе, помните, что все, что делает соединение более сильной кислотой, ослабит его конъюгат основания и наоборот . Задачи обзора строится 3.4 Структурные эффекты на кислотность и основность — Органическая химияМы узнали, что разные функциональные группы имеют разную кислотность.В этом разделе мы получим понимание фундаментальных причин этого, а именно: , почему одна группа более кислая, чем другая. Многие концепции, которые мы здесь изучим, будут по-прежнему применяться на протяжении всего курса, поскольку мы будем заниматься многими другими органическими темами. 3.4.1 Эффект элемента A. Периодический тренд: электроотрицательность Эффект элемента составляет около человека на ом, который соединяется с водородом (имейте в виду, что кислотность связана со способностью отдавать определенный водород).Давайте сравним кислотность водорода в этане, метиламине и этаноле, как показано ниже. Четкая тенденция в отношении кислотности этих соединений: кислотность увеличивается для элементов слева направо вдоль второй строки периодической таблицы, от C до N, а затем до O. Это согласуется с тенденцией к увеличению электроотрицательности вдоль период слева направо. Связь между электроотрицательностью и кислотностью можно объяснить тем, что атом с более высокой электроотрицательностью способен лучше приспособиться к отрицательному заряду сопряженного основания, поэтому лучше стабилизирует сопряженное основание.Следовательно, чем более стабильное основание конъюгата, тем слабее основание конъюгата и чем сильнее кислота . Что касается обсуждений в этом разделе, тенденция стабильности (или основности) конъюгированных оснований часто помогает объяснить тенденцию изменения кислотности. Относительная кислотность элементов за тот же период: Для элементов того же периода: чем электроотрицательнее атом, тем сильнее кислота; t Кислотность увеличивается слева направо в течение периода. B. Группа (по вертикали) Тренд: Размер атома При вертикальном движении в пределах данной группы периодической таблицы наблюдается тенденция к увеличению кислотности сверху вниз. Это можно проиллюстрировать с помощью галогенокислоты HX и галогенидов, как показано ниже: кислотность HX увеличивается сверху вниз, а основность конъюгированных оснований X — уменьшается сверху вниз. Кислотность H в тиоловой SH-группе также выше, чем у соответствующей спиртовой OH-группы, следуя той же тенденции.Например, p K a из CH 3 CH 2 SH составляет ~ 10, что намного более кислотно, чем этанол CH 3 CH 2 OH с ap K a из ~ 16. Чтобы понять эту тенденцию, мы еще раз рассмотрим устойчивость сопряженных оснований. При перемещении по вертикали в той же группе периодической таблицы, размер атома перекрывает его электроотрицательность в отношении основности. Атомный радиус йода примерно вдвое больше, чем у фтора, поэтому в иодид-ионе отрицательный заряд распространяется на значительно больший объем, поэтому I — более стабилен и менее щелочной, что делает HI более кислым. Относительная кислотность элементов в той же группе: Для элементов одной группы: чем больше размер атома, тем сильнее кислота; кислотность увеличивается сверху вниз по группе. 3.4.2. Резонансный эффект Резонансный эффект объясняет разницу в кислотности этанола и уксусной кислоты. И для этанола, и для уксусной кислоты водород связан с атомом кислорода, поэтому влияние элемента не имеет значения.Однако значения (и кислотность) этанола и уксусной кислоты сильно различаются. Что делает карбоновую кислоту намного более кислой, чем спирт? Как указывалось ранее, мы начинаем с рассмотрения стабильности конъюгированных оснований, помня, что более стабильное (более слабое) конъюгированное основание соответствует более сильной кислоте. Для ацетата, сопряженного основания уксусной кислоты, можно выделить два участника резонанса, и, следовательно, отрицательный заряд может быть делокализован (разделен) на два атома кислорода.Однако у этоксид-иона, сопряженного основания этанола, нет другого резонансного элемента, поэтому отрицательный заряд локализован на атоме кислорода. Как мы узнали в разделе 1.3 , разновидности, которые имеют больше вкладчиков резонанса, приобретают стабильность , поэтому ацетат более стабилен, чем этоксид, и слабее основания, поэтому уксусная кислота является более сильной кислотой, чем этанол. Делокализация заряда за счет резонанса оказывает очень сильное влияние на реакционную способность органических молекул, достаточную для объяснения большой разницы в более чем 10 p K a единиц между этанолом и уксусной кислотой.Поскольку p K a = –log K a , это означает, что существует коэффициент примерно 10 10 между значениями K a для двух молекул! p K a группы OH в спирте составляет около 15, однако OH в феноле (группа OH, соединенная с бензольным кольцом) имеет pK a около 10, что намного сильнее по кислотности, чем другие спирты. Объясните разницу. Решение : Разницу можно объяснить эффектом резонанса.Как упоминалось ранее, на конъюгированное основание этанола не наблюдается резонансного эффекта. Однако сопряженное основание фенола стабилизируется резонансным эффектом с еще четырьмя вкладчиками резонанса, а отрицательный элемент делокализован на бензольном кольце, поэтому сопряженное основание фенола намного более стабильно и является более слабым основанием. Следовательно, фенол намного более кислый, чем другие спирты.
3.4.3 Индуктивный эффект Давайте сравним значения p K a для уксусной кислоты и ее моно-, ди- и трихлорированных производных: Присутствие атомов хлора явно увеличивает кислотность группы карбоновой кислоты, и аргумент здесь, по-видимому, не имеет отношения к эффекту элемента.Эффект резонанса также не имеет к этому отношения, потому что для хлорированных молекул не может быть выявлено никаких дополнительных вкладчиков резонанса. Скорее, объяснение этого явления связано с так называемым индуктивным эффектом . Атом хлора более электроотрицателен, чем водород, и, таким образом, способен « индуцировать » или « притягивать » электронную плотность к себе через σ-связи между ними, и, следовательно, помогает распределить электронную плотность сопряженного основания, карбоксилата и стабилизируйте его.Заместитель хлора может упоминаться как электроноакцепторная группа из-за индуктивного эффекта. Эффект индукции — это эффект рассеивания заряда электроотрицательных атомов через σ-связи. Индуктивный эффект вызывает привыкание; большее количество атомов хлора имеет в целом более сильный эффект, что объясняет увеличение кислотности от моно-, ди- и трихлорированной уксусной кислоты. На следующей диаграмме в качестве примера показан индуктивный эффект трихлорацетата. Поскольку индукционный эффект зависит от электроотрицательности, заместители фтора обладают более сильным индуктивным действием, чем заместители хлора, что делает трифторуксусную кислоту (TFA) очень сильной органической кислотой. Кроме того, поскольку индукционный эффект осуществляется за счет ковалентных связей, его влияние значительно уменьшается с расстоянием — таким образом, хлор, находящийся на расстоянии двух атомов углерода от группы карбоновой кислоты, имеет более слабый эффект по сравнению с хлором, находящимся на расстоянии одного атома углерода. 3.4.4 Эффект гибридизации Чтобы представить эффект гибридизации, мы рассмотрим разницу в кислотности алкана, алкена и алкина. Атом водорода связан с атомом углерода во всех трех функциональных группах, поэтому эффект элемента не возникает. Также, учитывая сопряженное основание каждого, нет возможности дополнительного резонансного вкладчика. Ключевым различием между сопряженными анионами оснований является гибридизация атома углерода, то есть sp 3 , sp 2 и sp соответственно для алкана, алкена и алкина.Различные гибридизации приводят к разным s символу , то есть проценту s орбиталей от общего количества орбиталей. Гибридизация sp 3 означает 25% s символа (одна s и три p орбиталей, поэтому s символ составляет 1/4 = 25%), sp 2 гибридизация имеет 33,3% s характер, а число составляет 50% для sp гибридизации. Электроны на 2 s орбиталях находятся на более низком энергетическом уровне, чем электроны на 2 p орбиталях, потому что 2 s намного ближе к ядру.Таким образом, для аниона с характером более s электроны находятся ближе к ядру и испытывают более сильное притяжение, поэтому анион имеет меньшую энергию и более стабилен. Относительная стабильность трех анионов (сопряженных оснований) также может быть проиллюстрирована картой электростатического потенциала, на которой более светлый цвет (менее красный) указывает на меньшую электронную плотность аниона и более высокую стабильность. карта электростатических потенциалов конн. базы Это также может быть заявлено в более общем виде, что более символов в гибридных орбиталях делают атом более электроотрицательным. |