Хлорбензальмалондинитрил — Википедия
Материал из Википедии — свободной энциклопедии
У этого термина существуют и другие значения, см. CS.Хлорбензальмалонодинитрил (CS), «газ Сирень» — боевое отравляющее вещество, относящееся к группе ирритантов комплексного действия.
Хлорбензальмалонодинитрил был синтезирован в Миддлберском колледже двумя американцами Беном Корсоном (англ. Ben Corson) и Роджером Стаутоном (англ. Roger Stoughton) в 1928 году. Считается, что обозначение CS происходит от первых букв фамилий первооткрывателей.
CS — бесцветное, малолетучее кристаллическое вещество с запахом перца. Температура плавления 95 °C; температура кипения 310—315 °C. Плохо растворяется в воде (0,01 % при 30 °C), умеренно — в спирте, хорошо — в ацетоне, хлороформе. Вещество химически устойчиво, водой гидролизуется очень медленно с образованием о-хлорбензальдегида и малонодинитрила. В 95%-ном этаноле время гидролиза на 99 % составляет при 30°C 635 мин, при 40°С — 265 мин. Разбавленные щёлочи ускоряют гидролиз, кислоты замедляют его. Хлорбензальмалонодинитрил реагирует с окислителями с потерей раздражающих свойств. Термически устойчив до 300 °C, при 625 °C разлагается за 15-20 с.
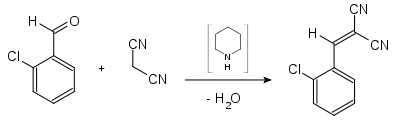
Получают хлорбензальмалонодинитрил по реакции, обратной гидролизу, в присутствии оснований (реакция Кнёвенагеля):
Боевое состояние — аэрозоль. Применяется с помощью химических авиационных бомб, артиллерийских снарядов, генераторов аэрозолей и дымовых гранат. CS в малых концентрациях обладает раздражающим действием на глаза и верхние дыхательные пути, а в больших концентрациях вызывает ожоги открытых участков кожи, в некоторых случаях — паралич дыхания, сердца и смерть. Признаки поражения: сильное жжение и боль в глазах и груди, сильное слезотечение, непроизвольное смыкание век, чихание, насморк (иногда с кровью), болезненное жжение во рту, носоглотке, в верхних дыхательных путях, кашель и боль в груди. При выходе из заражённой атмосферы или после надевания противогаза симптомы продолжают нарастать в течение 15 — 20 мин, а затем постепенно в течение 1 — 3 ч затихают. Защита от CS — противогаз, иногда требуются средства защиты кожи.
Непереносимая концентрация в воздухе 0,001—0,005 мг/л (1 мин). Поражающая концентрация 5 мг/м³. Непереносимая токсическая доза 0,02 г·мин/м³. Среднесмертельная токсическая доза (LCt50) 25 г·мин/м³. Поражающее действие на кожные покровы вдвое сильнее, чем у хлорацетофенона и бромбензилцианида.
Используется правоохранительными силами для разгона демонстраций и устранения уличных беспорядков, а также в газовом оружии самообороны: газовых баллончиках, патронах к газовым пистолетам и револьверам.
Хлорацетофенон — Википедия
Материал из Википедии — свободной энциклопедии
| Хлорацетофенон | |||
|---|---|---|---|
| |||
| Систематическое наименование | 2-хлоро-1-фенилэтанон | ||
| Традиционные названия | газ «Черёмуха» | ||
| Хим. формула | C8H7OCl | ||
| Состояние | твёрдое | ||
| Молярная масса | 154,59 г/моль | ||
| Плотность | 1,321 г/см³ | ||
| Энергия ионизации | 9,44 ± 0,01 эВ[1] | ||
| Температура | |||
| • плавления | 54-56 °C | ||
| • кипения | 247 °C | ||
| • вспышки | 244 ± 1 °F[1] | ||
| Давление пара | 0,09 гПа при 50°C | ||
| Растворимость | |||
| • в воде | 0,164 г/100 мл | ||
| Рег. номер CAS | 532-27-4 | ||
| PubChem | 10757 | ||
| Рег. номер EINECS | 208-531-1 | ||
| SMILES | |||
| InChI | |||
| RTECS | AM6300000 | ||
| ChemSpider | 10303 | ||
| ЛД50 | 17—81 мг/кг | ||
| Токсичность | высокотоксичен, ирритант, лакриматор | ||
| Пиктограммы ECB |  | ||
| NFPA 704 |  | ||
| Приведены данные для стандартных условий (25 °C, 100 кПа), если не указано иное. | |||
 (
(Хлорацетофено́н (CN, Литин, Орлит, Вещество № 34, Р-14) C6H5COCH2Cl — боевое отравляющее вещество из группы лакриматоров — слезоточивых веществ (ОВ раздражающего действия). Полицейское средство для разгона демонстрантов, захвата преступников и пр.; средство самообороны. В настоящее время из-за высокой токсичности постепенно вытесняется более безопасными ирритантами — CS, CR, OC, PAVA.
Армейские коды: CN (амер.), O-Salz (нем.), CAP (англ.), Grandite (фр.), ХАФ, «Черёмуха» (советск.).
Торговые названия: «Tear gas», Mace, Mace CN, Curb, Phaser, «Перец», «Гражданская оборона» и многие другие.
Другие химические названия:1-хлорацетофенон, 2-хлор-1-фенилэтанон, фенацилхлорид, фенилхлорметилкетон, хлорфенилметилкетон, α-хлорацетофенон.
Синтезирован немецким учёным Гребе в 1871 году[2]. В конце 1920-х годов впервые с успехом был применен французами при подавлении гражданских беспорядков в колониях и через несколько лет уже широко использовался полицией практически всех развитых стран. В 1923 г. правительство США финансировало масштабное исследование хлорацетофенона в Эджвудском арсенале. В годы Второй мировой войны были разработаны микропорошковые формы хлорацетофенона, обладавшие большей эффективностью и стойкостью на местности. В середине 1960-х на вооружение американской полиции поступил «мейс» — рецептура на основе раствора хлорацетофенона в керосине.
Белые кристаллы с запахом черёмухи или цветущих яблонь. Технический продукт имеет окраску от соломенно-жёлтой до серой. Нерастворим в воде, но хорошо растворяется в обычных органических растворителях — хлоралканах, сероуглероде, алифатических спиртах, эфирах, кетонах и в бензоле; в некоторых БОВ, например, иприте, фосгене, хлорпикрине и хлорциане. Термически стабилен, плавится и перегоняется без разложения. Устойчив к детонации.
Несмотря на низкую летучесть, пары хлорацетофенона делают местность непреодолимой без противогаза. Растворы хлорацетофенона в зависимости от плотности заражения, местных и метеорологических условий могут быть стойкими в течение часов и дней. Раствор хлорацетофенона в хлорпикрине в смеси с хлороформом (рецептура CNS) в летнее время в лесу стоек в течение 2 часов, а зимой даже до недели; на открытой местности летом примерно 1 час, а зимой 6 часов[3]
.По различным оценкам хлорацетофенон в 3—10 раз более токсичный, чем CS.
| Концентрация (мг/м³) | Действие |
|---|---|
| 0,05—0,3 | Минимальная концентрация, вызывающая в течение 10 с лёгкое раздражение глаз |
| 0,07—0,4 | При первом же вдохе лёгкое раздражение в носу |
| 0,1—0,7 | Порог восприятия запаха |
| 1,9 | Концентрации достаточная, чтобы разбудить спящего |
| 20—50 | ICt50 — концентрация выводящая из строя 50 % испытуемых (мг·мин/м³) |
| 7 000 | LCt50 — средняя смертельная концентрация (чистый аэрозоль, мг·мин/м³) |
| 14 000 | LCt50 — средняя смертельная концентрация (гранаты, мг·мин/м³) |
Хлорацетофенон — очень ядовит, типичный лакриматор, раздражение дыхательных путей выражено гораздо слабее чем при поражении CS и ОС. Начало действия через 0,5—2 мин. Продолжительность раздражающего действия 5—30 мин. Симптомы постепенно исчезают через 1—2 часа. Нахождение в облаке CN более 5 мин. считается опасным.
- Глаза: Слезотечение и резкая боль. При попадании растворов в глаза может вызывать ожог и помутнение роговицы, ослабление зрения.
- Дыхательные пути: пощипывание в носу, легкое жжение в горле, при высоких концентрациях — выделения из носа, боли в горле, возможны затрудненное дыхание, кашель.
- Кожа: Раздражающее действие, напоминающее ожог с образованием волдырей. Сильнее действует на влажную кожу. Вопреки распространённому мнению, хлорацетофенон гораздо более сильный кожный ирритант, чем CS. Накожная аппликация всего 0,5 мг CN в течение 60 мин. вызывает эритему у всех испытуемых. (для CS — не менее 20 мг).
- Военное применение. Наиболее эффективно применение хлорацетофенона в виде аэрозоля. Применяется в гранатах, генераторах аэрозолей (в том числе ранцевых), дымовых шашках и др.
- Применение органами правопорядка. Подразделения МВД РФ имеют в распоряжении различные виды гранат «Черёмуха», «Дрейф» и аэрозольный распылитель «Черёмуха-10М», содержащие хлорацетофенон.
- Применение гражданскими лицами. В РФ максимально разрешенное содержание хлорацетофенона в газовом баллончике — 80 мг, в газовых патронах — 100 мг.[4]. Импортные образцы могут содержать до 230 мг хлорацетофенона на патрон. Цветовая маркировка патрона — голубая, синяя. В настоящее время практически полностью вытеснен с рынка средствами самообороны на основе CS, CR, OC.
Для защиты от поражения парами или аэрозолем хлорацетофенона достаточно надеть противогаз.
Советский войсковой прибор химической разведки (ВПХР) способен определить хлорацетофенон в концентрации 0,002—0,2 мг/л.
Для дегазации применяют подогретые водно-спиртовые растворы сульфида натрия
- ↑ 1 2 http://www.cdc.gov/niosh/npg/npgd0119.html
- ↑ Graebe, C. Über eine neue Klasse von Alkoholen. Berichte, 1871, 4, 34-35
- ↑ Франке З. Химия отравляющих веществ. — М.:Химия, 1973. − Т.1. − 440 с.
- ↑ ГОСТ Р 50742-95 Государственный стандарт РФ. Патроны к газовым пистолетам и револьверам
- ↑ Александров В. Н., Емельянов В. И. Отравляющие вещества. — М:Воениздат,1990. — С. 214—217. ISBN 5-203-00341-6
Хром — Википедия
| Хром | |
|---|---|
| ← Ванадий | Марганец → | |
| Твёрдый металл голубовато-белого цвета | |
 | |
| Название, символ, номер | Хром / Chromium (Cr), 24 |
| Атомная масса (молярная масса) | 51,9961(6)[1] а. е. м. (г/моль) |
| Электронная конфигурация | [Ar] 3d5 4s1 |
| Радиус атома | 130 пм |
| Ковалентный радиус | 118 пм |
| Радиус иона | (+6e)52 (+3e)63 пм |
| Электроотрицательность | 1,66 (шкала Полинга) |
| Электродный потенциал | −0,74 |
| Степени окисления | 6, 3, 2, 0 |
| Энергия ионизации (первый электрон) | 652,4 (6,76) кДж/моль (эВ) |
| Плотность (при н. у.) | 7,19 г/см³ |
| Температура плавления | 2130 K (1856,9 °C) |
| Температура кипения | 2945 K (2671,9 °C) |
| Уд. теплота плавления | 21 кДж/моль |
| Уд. теплота испарения | 342 кДж/моль |
| Молярная теплоёмкость | 23,3[2] Дж/(K·моль) |
| Молярный объём | 7,23 см³/моль |
| Структура решётки | кубическая объёмноцентрированая |
| Параметры решётки | 2,885 Å |
| Температура Дебая | 460 K |
| Теплопроводность | (300 K) 93,9 Вт/(м·К) |
| Номер CAS | 7440-47-3 |
Хром — элемент 6-й группы (по устаревшей классификации — побочной подгруппы 6-й группы) 4-го периода периодической системы химических элементов Д. И. Менделеева с атомным номером 24. Обозначается символом Cr (лат. Chromium). Простое вещество хром — твёрдый металл голубовато-белого цвета. Хром иногда относят к чёрным металлам.
Происхождение названия[править | править код]
Название элемент получил от греч. χρῶμα — цвет, краска — из-за разнообразия окраски своих соединений.
История[править | править код]
Открыт на Среднем Урале, в Березовском золоторудном месторождении. Впервые упоминается в труде М. В. Ломоносова «Первые основания металлургии» (1763 год), как красная свинцовая руда, PbCrO4. Современное название — крокоит. В 1797 году французский химик Л. Н. Воклен выделил из него новый тугоплавкий металл ( Воклен получил карбид хрома). Он прокалил зелёный оксид Cr2O3 с углём и выделил тугоплавкий металл (с примесью карбидов). Сам оксид Cr2O3 Воклен получил разложением «Сибирского красного свинца» — минерала крокоита PbCrO4.
Современный способ получения чистого хрома (с 1894 г.) отличается от способа Воклена только видом восстановителя. Процесс электролитического покрытия железа хромом разработан в 20-х годах ХХ в.
Хром является довольно распространённым элементом в земной коре (0,03 % по массе)[3]. Основные соединения хрома — хромистый железняк (хромит) FeO·Cr2O3. Вторым по значимости минералом является крокоит PbCrO4.
Месторождения[править | править код]
Самые большие месторождения хрома находятся в ЮАР (1-е место в мире), Казахстане, России, Зимбабве, Мадагаскаре. Также есть месторождения на территории Турции, Индии, Армении[4], Бразилии, на Филиппинах[5].
Главные месторождения хромовых руд в РФ известны на Урале (Донские и Сарановское).
Разведанные запасы в Казахстане составляют свыше 350 миллионов тонн (2-е место в мире)[5].
Среднее содержание хрома в различных изверженных породах резко непостоянно. В ультраосновных породах (перидотитах) оно достигает 2 кг/т, в основных породах (базальтах и др.) — 200 г/т, а в гранитах десятки г/т. Кларк хрома в земной коре 83 г/т. Он является типичным литофильным элементом и почти весь заключен в минералах типа хромшпинелидов. Хром вместе с железом, титаном, никелем, ванадием и марганцем составляют одно геохимическое семейство.
Различают три основных минерала хрома: магнохромит (Mg, Fe)Cr2O4, хромпикотит (Mg, Fe)(Cr, Al)2O4 и алюмохромит (Fe, Mg)(Cr, Al)2O4. По внешнему виду они неразличимы, и их неточно называют «хромиты». Состав их изменчив:
- Cr2O3 18—62 %,
- FeO 1—18 %,
- MgO 5—16 %,
- Al2O3 0,2 — 0,4 (до 33 %),
- Fe2O3 2 — 30 %,
- примеси TiO2 до 2 %,
- V2O5 до 0,2 %,
- ZnO до 5 %,
- MnO до 1 %; присутствуют также Co, Ni и др.
Собственно, хромит, то есть FeCr2O4 сравнительно редок. Помимо различных хромитов, хром входит в состав ряда других минералов — хромовой слюды (фуксита), хромового хлорита, хромвезувиана, хромдиопсида, хромтурмалина, хромового граната (уваровита) и др., которые нередко сопровождают руды, но сами промышленного значения не имеют. В экзогенных условиях хром, как и железо, мигрирует в виде взвесей и может накапливаться в глинах. Наиболее подвижной формой являются хроматы.
Хром встречается в природе в основном в виде хромистого железняка Fe(CrO2)2 (хромит железа). Из него получают феррохром восстановлением в электропечах коксом (углеродом):
- Fe(CrO2)2+4C→Fe+2Cr+4CO{\displaystyle {\mathsf {Fe(CrO_{2})_{2}+4C\rightarrow Fe+2Cr+4CO}}}
Феррохром применяют для производства легированных сталей.
Чтобы получить чистый хром, реакцию ведут следующим образом:
1) сплавляют хромит железа с карбонатом натрия (кальцинированная сода) на воздухе:
- 4Fe(CrO2)2+8Na2CO3+7O2→8Na2CrO4+2Fe2O3+8CO2{\displaystyle {\mathsf {4Fe(CrO_{2})_{2}+8Na_{2}CO_{3}+7O_{2}\rightarrow 8Na_{2}CrO_{4}+2Fe_{2}O_{3}+8CO_{2}}}}
2) растворяют хромат натрия и отделяют его от оксида железа;
3) переводят хромат в дихромат, подкисляя раствор и выкристаллизовывая дихромат;
4) получают чистый оксид хрома восстановлением дихромата натрия углём:
- Na2Cr2O7+2C→Cr2O3+Na2CO3+CO{\displaystyle {\mathsf {Na_{2}Cr_{2}O_{7}+2C\rightarrow Cr_{2}O_{3}+Na_{2}CO_{3}+CO}}}
5) с помощью алюминотермии получают металлический хром:
- Cr2O3+2Al→Al2O3+2Cr+130kcal{\displaystyle {\mathsf {Cr_{2}O_{3}+2Al\rightarrow Al_{2}O_{3}+2Cr+130kcal}}}
6) с помощью электролиза получают электролитический хром из раствора хромового ангидрида в воде, содержащего добавку серной кислоты. При этом на катодах совершаются в основном 3 процесса:
- восстановление шестивалентного хрома до трехвалентного с переходом его в раствор;
- разряд ионов водорода с выделением газообразного водорода;
- разряд ионов, содержащих шестивалентный хром, с осаждением металлического хрома;
- Cr2O72−+14H++12e−→2Cr+7h3O{\displaystyle {\mathsf {Cr_{2}O_{7}^{2-}+14H^{+}+12e^{-}\rightarrow 2Cr+7H_{2}O}}}
В свободном виде — голубовато-белый металл с кубической объёмноцентрированной решёткой, a = 0,28845 нм. Ниже температуры 38 °C является антиферромагнетиком, выше переходит в парамагнитное состояние (точка Нееля).
Хром имеет твёрдость по шкале Мооса 8.5[6], один из самых твёрдых чистых металлов (уступает только иридию, бериллию, вольфраму и урану). Очень чистый хром достаточно хорошо поддаётся механической обработке.
Изотопы[править | править код]
Известны изотопы хрома с массовыми числами от 42 до 67 (количество протонов 24, нейтронов от 18 до 43) и 2 ядерных изомера.
Природный хром состоит из четырех стабильных изотопов (50Cr (изотопная распространённость 4,345 %), 52Cr (83.789 %), 53Cr (9.501 %), 54Cr (2.365 %)).
Среди искусственных изотопов самый долгоживущий 51Cr (период полураспада 27 суток). Период полураспада остальных не превышает одних суток.
Характерные степени окисления[править | править код]
Для хрома характерны степени окисления +2, +3 и +6 (см. табл.). Практически все соединения хрома окрашены[7].
| Степень окисления | Оксид | Гидроксид | Характер | Преобладающие формы в растворах | Примечания |
|---|---|---|---|---|---|
| +2 | CrO (чёрный) | Cr(OH)2 (жёлтый) | Основный | Cr2+ (соли голубого цвета) | Очень сильный восстановитель |
| +3 | Cr2O3 (зелёный) | Cr(OH)3 (серо-зелёный) | Амфотерный | Cr3+ (зелёные или лиловые соли) [Cr(OH)4]− (зелёный) | |
| +4 | CrO2 | не существует | Несолеобразующий | — | Встречается редко, малохарактерна |
| +6 | CrO3 (красный) | H2CrO4 H2Cr2O7 | Кислотный | CrO42− (хроматы, желтые) Cr2O72− (дихроматы, оранжевые) | Переход зависит от рН среды. Сильнейший окислитель, гигроскопичен, очень ядовит. |

Простое вещество[править | править код]
Устойчив на воздухе за счёт пассивирования. По этой же причине не реагирует с серной и азотной кислотами. При 2000 °C сгорает с образованием зелёного оксида хрома(III) Cr2O3, обладающего амфотерными свойствами.
Синтезированы соединения хрома с бором (бориды Cr2B, CrB, Cr3B4, CrB2, CrB4 и Cr5B3), с углеродом (карбиды Cr23C6, Cr7C3 и Cr3C2), c кремнием (силициды Cr3Si, Cr5Si3 и CrSi) и азотом (нитриды CrN и Cr2N).
Соединения Cr(+2)[править | править код]
Степени окисления +2 соответствует основный оксид CrO (чёрный). Соли Cr2+ (растворы голубого цвета) получаются при восстановлении солей Cr3+ или дихроматов цинком в кислой среде («водородом в момент выделения»):
- 2Cr3+→Zn,HCl[H]2Cr2+{\displaystyle {\mathsf {2Cr^{3+}{\xrightarrow[{Zn,HCl}]{[H]}}2Cr^{2+}}}}
Все эти соли Cr2+ — сильные восстановители вплоть до того, что при стоянии вытесняют водород из воды[8]. Кислородом воздуха, особенно в кислой среде, Cr2+ окисляется, в результате чего голубой раствор быстро зеленеет.
Коричневый или жёлтый гидроксид Cr(OH)2 осаждается при добавлении щелочей к растворам солей хрома(II).
Синтезированы дигалогениды хрома CrF2, CrCl2, CrBr2 и CrI2
Соединения Cr(+3)[править | править код]
Степени окисления +3 соответствует амфотерный оксид Cr2O3 и гидроксид Cr(OH)3 (оба — зелёного цвета). Это — наиболее устойчивая степень окисления хрома. Соединения хрома в этой степени окисления имеют цвет от грязно-лилового (в водных растворах ион Cr3+ существует в виде аквакомплексов [Cr(H2O)6]3+) до зелёного (в координационной сфере присутствуют анионы).
Cr3+ склонен к образованию двойных сульфатов вида MICr(SO4)2·12H2O (квасцов)
Гидроксид хрома (III) получают, действуя аммиаком на растворы солей хрома (III):
- Cr3++3Nh4+3h3O→Cr(OH)3↓+3Nh5+{\displaystyle {\mathsf {Cr^{3+}+3NH_{3}+3H_{2}O\rightarrow Cr(OH)_{3}\downarrow +3NH_{4}^{+}}}}
Можно использовать растворы щелочей, но в их избытке образуется растворимый гидроксокомплекс:
- Cr3++3OH−→Cr(OH)3↓{\displaystyle {\mathsf {Cr^{3+}+3OH^{-}\rightarrow Cr(OH)_{3}\downarrow }}}
- Cr(OH)3+3OH−→[Cr(OH)6]3−{\displaystyle {\mathsf {Cr(OH)_{3}+3OH^{-}\rightarrow [Cr(OH)_{6}]^{3-}}}}
Сплавляя Cr2O3 со щелочами, получают хромиты:
- Cr2O3+2NaOH→2NaCrO2+h3O{\displaystyle {\mathsf {Cr_{2}O_{3}+2NaOH\rightarrow 2NaCrO_{2}+H_{2}O}}}
Непрокаленный оксид хрома(III) растворяется в щелочных растворах и в кислотах:
- Cr2O3+6HCl→2CrCl3+3h3O{\displaystyle {\mathsf {Cr_{2}O_{3}+6HCl\rightarrow 2CrCl_{3}+3H_{2}O}}}
При окислении соединений хрома(III) в щелочной среде образуются соединения хрома(VI):
- 2Na3[Cr(OH)6]+3h3O2→2Na2CrO4+2NaOH+8h3O{\displaystyle {\mathsf {2Na_{3}[Cr(OH)_{6}]+3H_{2}O_{2}\rightarrow 2Na_{2}CrO_{4}+2NaOH+8H_{2}O}}}
То же самое происходит при сплавлении оксида хрома (III) со щелочью и окислителями, или со щелочью на воздухе (расплав при этом приобретает жёлтую окраску):
- 2Cr2O3+8NaOH+3O2→4Na2CrO4+4h3O{\displaystyle {\mathsf {2Cr_{2}O_{3}+8NaOH+3O_{2}\rightarrow 4Na_{2}CrO_{4}+4H_{2}O}}}
Соединения хрома (+4)[править | править код]
При осторожном разложении оксида хрома(VI) CrO3 в гидротермальных условиях получают оксид хрома(IV) CrO2, который является ферромагнетиком и обладает металлической проводимостью.
Среди тетрагалогенидов хрома устойчив CrF4, тетрахлорид хрома CrCl4 существует только в парах.
Соединения хрома (+6)[править | править код]
Степени окисления +6 соответствует кислотный оксид хрома (VI) CrO3 и целый ряд кислот, между которыми существует равновесие. Простейшие из них — хромовая H2CrO4 и двухромовая H2Cr2O7. Они образуют два ряда солей: желтые хроматы и оранжевые дихроматы соответственно.
Оксид хрома (VI) CrO3 образуется при взаимодействии концентрированной серной кислоты с растворами дихроматов. Типичный кислотный оксид, при взаимодействии с водой он образует сильные неустойчивые хромовые кислоты: хромовую H2CrO4, дихромовую H2Cr2O7 и другие изополикислоты с общей формулой H2CrnO3n+1. Увеличение степени полимеризации происходит с уменьшением рН, то есть увеличением кислотности:
- 2CrO42−+2H+→Cr2O72−+h3
Фосген — Википедия
| Фосген | |
|---|---|
 ( {{{картинка}}}) | |
| Систематическое наименование | Дихлорид карбонила |
| Традиционные названия | Фосген |
| Хим. формула | COCl2 |
| Состояние | бесцветный газ с неприятным запахом |
| Молярная масса | 98,92 г/моль |
| Плотность | 4,248 кг/м³ |
| Энергия ионизации | 11,55 ± 0,01 эВ[1] |
| Температура | |
| • плавления | −118 °C |
| • кипения | +8,3 °C |
| Давление пара | 1,6 ± 0,1 атм[1] |
| Дипольный момент | 1,17 Д |
| Рег. номер CAS | 75-44-5 |
| PubChem | 6371 |
| Рег. номер EINECS | 200-870-3 |
| SMILES | |
| InChI | |
| RTECS | SY5600000 |
| ChEBI | 29365 |
| Номер ООН | 1076 |
| ChemSpider | 6131 |
| ЛД50 | 0,334 мг/л*10 мин. (LC50, крыса, ингаляция) |
| Токсичность | чрезвычайно токсичен, обладает сильным удушающим действием. |
| Фразы риска (R) | R26, R34 |
| Фразы безопасности (S) | (S1/2), S9, S26, S36/37/39, S45 |
| Краткие характер. опасности (H) | h430, h414, h380, EUH071 |
| Меры предостор. (P) | P260, P280, P304+P340, P303+P361+P353, P305+P351+P338, P315, P405, P403 |
| Сигнальное слово | ОПАСНО! |
| Пиктограммы СГС |     |
| NFPA 704 | |
| Приведены данные для стандартных условий (25 °C, 100 кПа), если не указано иное. | |
Фосге́н (дихлорангидрид угольной кислоты) — химическое вещество с формулой COCl2, при нормальных условиях — бесцветный чрезвычайно токсичный и удушливый газ с запахом прелого сена. Синонимы: оксид-дихлорид углерода, карбонилхлорид, хлорокись углерода.
Обладает удушающим действием. Использовался в Первую мировую войну как боевое отравляющее вещество.
tкип= +8,2 °C, tпл= −118 °C, плотность в жидкой фазе 1,403 г/см³ (при температуре кипения), в газовой фазе 4,248 кг/м³ (+15 °C, 1 бар)[2]; плохо растворим в воде, хорошо — в органических растворителях.
Фосген представляет собой бесцветный газ, который ниже +8,2 °C конденсируется в бесцветную жидкость. Его запах напоминает прелые фрукты или сено. Технический продукт имеет слегка желтоватую или красновато-жёлтую окраску. Фосген примерно в 3,5 раза тяжелее воздуха. Из-за высокого давления пара он даже при низких температурах обладает большой летучестью. Фосген можно легко конденсировать сжатием, его критическая температура составляет 183 °C, критическое давление 56 кгс/см². В холодной воде фосген растворим мало −0,9 %. Он легко растворим в органических растворителях, например в бензине, толуоле, ксилоле, уксусной кислоте, хлороформе.
При обычной температуре фосген — стабильное соединение. При сильном нагревании он частично разлагается на хлор и окись углерода. Выше 800 °C он полностью диссоциирует. Количество ядовитых продуктов разложения при взрыве ничтожно, поэтому возможно применение фосгена во взрывных боеприпасах.
При хранении фосгена в стальных ёмкостях, например при длительном нахождении в минах, образуется пентакарбонил железа Fe(CO)5. Это — красновато-жёлтая жидкость, тяжелее фосгена, и разлагаемая на свету фотокаталитически с образованием ядовитой окиси углерода. Фосген почти не гидролизуется парами воды, поэтому концентрация фосгена в воздухе, заметно падает лишь через длительное время. При высокой влажности воздуха облако фосгена за счёт частичного гидролиза может приобрести беловатый оттенок.
Энергично реагирует с аммиаком с образованием карбамида и хлорида аммония:
- COCl2+4Nh4→(Nh3)2CO+2Nh5Cl.{\displaystyle {\mathsf {COCl_{2}+4NH_{3}\rightarrow (NH_{2})_{2}CO+2NH_{4}Cl}}.}
Эта реакция используется для экспресс-обнаружения утечек фосгена — смоченный водным раствором аммиака ватный тампон в присутствии фосгена начинает заметно выделять белый дым состоящий из кристалликов хлорида аммония. Обнаружению фосгена этим способом мешает присутствие хлора, который с аммиаком также образует дым хлорида аммония.
Впервые фосген получил Гемфри Дэви в 1812 году путём облучения солнечным светом смеси хлора с окисью углерода[3].
- CO+Cl2→hνCOCl2.{\displaystyle {\mathsf {CO+Cl_{2}}}{\xrightarrow {h\nu }}{\mathsf {COCl_{2}}}.}
Дэви назвал образовавшееся вещество «фосген» (англ. phosgen, букв. «светорождённый», от др.-греч. φῶς «свет» и γίγνομαι «порождаю»)[3].
Фосген образуется также при окислении хлороформа кислородом воздуха под действием света:
- 2CHCl3+O2 →hν 2COCl2+2HCl{\displaystyle {\mathsf {2CHCl_{3}+O_{2}}}\ {\xrightarrow {h\nu }}\ {\mathsf {2COCl_{2}+2HCl}}}
В промышленности получают нагреванием СО с Cl2 в присутствии катализатора:
- CO+Cl2 →150oC,C/Pt COCl2.{\displaystyle {\mathsf {CO+Cl_{2}\ {\xrightarrow {150^{o}C,C/Pt}}\ COCl_{2}}}.}
В лаборатории может быть легко получен несильным нагреванием смеси CCl4 и SO3 (или олеума):
- 2SO3+CCl4→S2O5Cl2+COCl2.{\displaystyle {\mathsf {2SO_{3}+CCl_{4}\rightarrow S_{2}O_{5}Cl_{2}+COCl_{2}}}.}
Также фосген образуется при горении некоторых хлорсодержащих фреонов, вследствие чего запрещено курение при обслуживании холодильных машин и установок.
Обладает удушающим действием. Смертельная концентрация 0,01—0,03 мг/л (при экспозиции 15 минут). Контакт фосгена с лёгочной тканью вызывает нарушение проницаемости альвеол и быстро прогрессирующий отёк лёгких. Антидот неизвестен.
Токсические свойства[править | править код]
Фосген очень ядовит, но только при вдыхании паров. Первые отчётливые признаки отравления появляются после скрытого периода от 4 до 8 часов; наблюдались даже периоды в 15 часов.
По различным данным вдыхание фосгена в концентрации 0,004 мг/л в течение 60—90 минут не приводит к отравлению.
Пребывание в атмосфере, содержащей до 0,01 мг/л фосгена, возможно максимально в течение 1 часа. При этом восприимчивые люди уже могут получить лёгкое отравление. Концентрации в 0,022 мг/л являются смертельными уже через 30 минут воздействия. В 50 % случаев отравление при вдыхании 0,1 мг/л в течение 30—60 минут приводит к смерти. Остальные 50 % оставшихся в живых длительно небоеспособны в результате тяжелейших отравлений. Даже при малом времени воздействия таких концентраций могут произойти сильные отравления, иногда заканчивающиеся смертью.
Концентрация 1 мг/л при времени экспозиции 5 минут в 50—75 % случаев отравления ведёт к смерти; меньшие концентрации (0,5—0,8 мг/л) приводят к тяжёлым отравлениям.
Концентрация 5 мг/л смертельна уже через 2—3 секунды[источник не указан 76 дней].
Малые концентрации фосгена влияют на вкусовые ощущения. Так, например, курить сигарету в содержащем фосген воздухе неприятно или вовсе невозможно.
Физиологическое действие[править | править код]
Токсический отёк лёгких, возникающий после вдыхания паров фосгена, дифосгена, трифосгена, проявляется лишь после скрытого периода в несколько часов. В этот период отравленный чувствует себя хорошо, и как правило вполне дееспособен. У восприимчивых людей в это время появляется сладкий, часто противный привкус во рту, иногда тошнота и рвота. В большинстве случаев возникают незначительные позывы к кашлю, першение и жжение в носоглотке, небольшие нарушения ритма дыхания и пульса.
После латентного периода наступает сильный кашель, одышка, синюшность лица и губ.
Прогрессирующий отёк лёгких ведёт к сильному удушью, мучительному давлению в грудной клетке, ритм дыхания увеличивается от 18—20 в минуту (норма) до 30—50 в минуту, в кризисе — до 60—70 в минуту. Дыхание судорожное. Содержащая белок отёчная пенистая и вязкая жидкость выбрызгивается из альвеол и бронхиол в более широкие дыхательные пути, ведёт к затруднению и невозможности дыхания. Отравленный отхаркивает большие количества этой жидкости, часто смешанной с кровью. При токсическом отёке лёгких примерно до половины общего количества крови организма переходит в лёгкие, которые в результате этого опухают и увеличиваются в массе. В то время как нормальное лёгкое весит около 500—600 грамм, можно было наблюдать «фосгеновые» лёгкие весом до 2,5 килограмм.
Кровяное давление резко падает, отравленный пребывает в сильнейшем возбуждении, дышит с шумом, хватает ртом воздух, затем наступает смерть.
Встречаются также случаи, когда отравленный избегает любого лишнего движения и для облегчения дыхания выбирает какое-то наиболее удобное положение. Губы у таких отравленных серые, пот холодный и липкий. Несмотря на удушье, мокрота у них не отделяется. Через несколько дней отравленный умирает.
Редко, через 2—3 суток может наступить улучшение состояния, которое через 2—3 недели может закончиться выздоровлением, но часты осложнения в результате вторичных инфекционных заболеваний, что приводит к смертельному исходу.
При очень высоких концентрациях отёк лёгких не развивается. Отравленный делает глубокие вдохи, падает на землю, корчится и бьётся в судорогах, кожа на лице становится от фиолетово-синей до тёмно-синей, и очень быстро наступает смерть.
Хеглер на примере одного поражения так описывает коварный характер отравления фосгеном:
Сильный и здоровый юноша 19 лет случайно попал в облако фосгена, распространявшегося по реке. Он поспешил выйти из атмосферы с непривычным запахом и быстро причалил к берегу. Затем юноша обратился к врачу по поводу возникшего у него кашля. Врач не смог обнаружить никаких симптомов заболевания, хотя обследовал пострадавшего очень тщательно. Следуя совету врача, молодой человек для устранения незначительного недомогания пошёл прогуляться. Однако уже через 4 часа он был доставлен в больницу с сильным отёком лёгких, при сильнейшем цианозе, но пока ещё с нормальной деятельностью сердца. В процессе госпитализации через 4,5 часа после отравления наступила смерть.
Один из известных токсикологов Мунтш так описывал состояние поражённого фосгеном человека:
Сильнейшей степени достигает цианоз и одышка; больные стонут и просят воздуха. Умирающий как бы тонет в собственной жидкости, постепенно заполняющей лёгкие….
Охрана труда[править | править код]
Фосген очень токсичен[4]. Запах фосгена ощутим в концентрации 0,004 мг/л, однако на обонятельный нерв фосген влияет так, что в дальнейшем обоняние притупляется и перестают ощущаться даже более высокие концентрации[5]. При опасной концентрации люди могут не почувствовать запах фосгена[6]; его ПДК в воздухе рабочей зоны равна 0,5 мг/м3 (максимально-разовая)[7]. А порог восприятия запаха может быть, например, 4 мг/м3[8].
Можно ожидать, что использование широко распространённых фильтрующих СИЗОД в сочетании с «заменой фильтров по появлении запаха под маской» (как это почти всегда рекомендуется в РФ поставщиками СИЗОД) приведёт к чрезмерному воздействию фосгена на, по крайней мере, часть работников — из-за запоздалой замены противогазных фильтров. Для защиты от фосгена следует использовать значительно более эффективные изменение технологии и средства коллективной защиты.
Использование в качестве боевого отравляющего вещества[править | править код]
Использовался в Первую мировую войну как боевое отравляющее вещество.

Летучесть фосгена достаточна для достижения токсических концентраций в зимнее время. Стойкость при −20 °C составляет около трёх часов, в летние месяцы она чрезвычайно мала — не более 30 минут. Летучесть при −20 °C равна 1,4 г/л, при +20 °C — около 6,4 г/л. Вследствие обычных метеорологических воздействий фактическая концентрация фосгена в воздухе меньше и едва ли превышает 1 г/л.
С военной точки зрения представляет интерес хорошая растворимость фосгена в хлорпикрине, иприте, арил- и алкилхлорарсинах и в кислотных дымообразователях — четырёххлористых кремнии, олове, титане. Смеси фосгена с дымообразователями применялись в Первую мировую войну и были запасены в больших количествах во время Второй мировой войны.
Военные обозначения[править | править код]
Использование в органическом синтезе[править | править код]
Очень активен во многих реакциях присоединения, благодаря этому активно используется в органическом синтезе (фосгенирование). Применяется для получения ряда красителей.
Методом межфазной поликонденсации раствора фосгена в метиленхлориде с щелочным раствором 2,2-бис(4-оксифенил)пропана (более известен как бисфенол — А) в присутствии катализатора получают один из важных термопластов инженерно-технического назначения — поликарбонат.
- Химическая энциклопедия / Редкол.: Кнунянц И. Л. и др. — М.: Советская энциклопедия, 1995. — Т. 4 (Пол-Три). — 639 с. — ISBN 5-82270-092-4.
Арсин — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 31 января 2016; проверки требуют 29 правок. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 31 января 2016; проверки требуют 29 правок.| Арсин | |||
|---|---|---|---|
 ( ({{{картинка малая}}}) | |||
| Систематическое наименование | арсин | ||
| Традиционные названия | мышьяковистый водород | ||
| Хим. формула | AsH3 | ||
| Состояние | бесцветный газ | ||
| Молярная масса | 77.95 г/моль | ||
| Плотность | 4.93 г/л, газ; 1.640 г/мл (−64 °C) | ||
| Энергия ионизации | 9,89 ± 0,01 эВ[1] | ||
| Температура | |||
| • плавления | −117 °C | ||
| • кипения | −62.5 °C | ||
| Пределы взрываемости | 5,1 ± 0,1 об.%[1] | ||
| Энтальпия | |||
| • образования | +66.4 кДж/моль | ||
| Давление пара | 14,9 ± 0,1 атм[1] | ||
| Растворимость | |||
| • в воде | 0.07 г/100 мл | ||
| Дипольный момент | 0.20 Д | ||
| Рег. номер CAS | 7784-42-1 | ||
| PubChem | 23969 | ||
| Рег. номер EINECS | 232-066-3 | ||
| SMILES | |||
| InChI | |||
| RTECS | CG6475000 | ||
| ChEBI | 47217 | ||
| Номер ООН | 2188 | ||
| ChemSpider | 22408 | ||
| Токсичность | Чрезвычайно ядовит, СДЯВ | ||
| Фразы риска (R) | R26 | ||
| Фразы безопасности (S) | S20/21, S28, S36/37 | ||
| Краткие характер. опасности (H) | |||
| Меры предостор. (P) | P261, P301+P310, P321, P304+P340, P405, P501 | ||
| Сигнальное слово | Опасно | ||
| Пиктограммы СГС |   | ||
| NFPA 704 | | ||
| Приведены данные для стандартных условий (25 °C, 100 кПа), если не указано иное. | |||

 (
(Арси́н (мышьяковистый водород, арсенид водорода) — вещество с формулой AsH3 (правильнее H3As), химическое соединение мышьяка и водорода. При нормальных условиях — очень токсичный бесцветный газ. Абсолютно химически чистый арсин запаха не имеет, но ввиду неустойчивости продукты его окисления придают арсину чесночный запах. Открыт шведским химиком Карлом Вильгельмом Шееле в 1775 году.
Молекула арсина имеет форму тригональной пирамиды с атомом мышьяка в вершине. Низкое значение дипольного момента, которое составляет 0,20D, свидетельствует, что связь в молекуле арсина близка к неполярной и арсин практически не проявляет электронодонорные свойства. Так, ион арсония AsH4+, в отличие от его аналога иона аммония NH4+ и даже фосфония PH4+ неустойчив и был обнаружен лишь спектроскопически при пониженной температуре.
В промышленности получают гидролизом арсенидов металлов (Mg, Zn и др.) кислотами или восстановлением соединений мышьяка водородом, взаимодействием галогенидов мышьяка с Li[AlH4], Na[BH4] или другими гидридами, например:
- Na3As+3h3O→Ash4↑+3NaOH{\displaystyle {\mathsf {Na_{3}As+3H_{2}O\rightarrow AsH_{3}\uparrow +3NaOH}}}
- As2O3+6Zn+6h3SO4→2Ash4↑+6ZnSO4+3h3O{\displaystyle {\mathsf {As_{2}O_{3}+6Zn+6H_{2}SO_{4}\rightarrow 2AsH_{3}\uparrow +6ZnSO_{4}+3H_{2}O}}}
- Ash4+6AgNO3+3h3O→6Ag↓+As(OH)3+6HNO3{\displaystyle {\mathsf {AsH_{3}+6AgNO_{3}+3H_{2}O\rightarrow 6Ag\downarrow +As(OH)_{3}+6HNO_{3}}}}
- Арсин сравнительно нестоек и медленно разлагается даже при комнатной температуре на водород и элементарный мышьяк, при температуре 500 °C — мгновенно:
- 2Ash4→2As+3h3{\displaystyle {\mathsf {2AsH_{3}\rightarrow 2As+3H_{2}}}}
- При пропускании AsH3 через нагретую, наполненную водородом стеклянную трубку, металлический мышьяк отлагается на стенках трубки в виде черно-бурого зеркала. На этом свойстве арсина основана высокочувствительная качественная реакция на мышьяк — проба Марша.
- Не самовоспламеняется на воздухе и в кислороде при комнатной температуре, но при нагревании на воздухе до 200 °C сгорает:
- 2Ash4+3O2→As2O3+3h3O{\displaystyle {\mathsf {2AsH_{3}+3O_{2}\rightarrow As_{2}O_{3}+3H_{2}O}}}
- В хлоре самовоспламеняется даже при −196оС, с замещением водорода на хлор и выделением хлороводорода:
- Ash4+3Cl2→AsCl3+3HCl{\displaystyle {\mathsf {AsH_{3}+3Cl_{2}\rightarrow AsCl_{3}+3HCl}}}
- С бромом и йодом реагирует таким же образом, давая соответствующие галогениды.
- 2Ash4+3S→3h3S+2As{\displaystyle {\mathsf {2AsH_{3}+3S\rightarrow 3H_{2}S+2As}}}
- Ash4+3HCl→AsCl3+3h3{\displaystyle {\mathsf {AsH_{3}+3HCl\rightarrow AsCl_{3}+3H_{2}}}}
- Реагирует с растворами щелочных металлов в жидком аммиаке, проявляя кислотные свойства и образуя мышьяковистые производные, аналогичные амидам щелочных металлов:
- Ash4+NaNh3→NaAsh3+Nh4{\displaystyle {\mathsf {AsH_{3}+NaNH_{2}\rightarrow NaAsH_{2}+NH_{3}}}}
- При нагревании арсина с металлами образуются арсениды.
При взаимодействии хлорида мышьяка AsCl3 с диметилцинком образуются соответствующие органические производные арсина, например, триметиларсин:
- 2AsCl3+3Zn(Ch4)2→2As(Ch4)3+3ZnCl2{\displaystyle {\mathsf {2AsCl_{3}+3Zn(CH_{3})_{2}\rightarrow 2As(CH_{3})_{3}+3ZnCl_{2}}}}
Это ядовитые жидкости с отвратительным запахом, проявляющие свойства ненасыщенных соединений.
Токсичность[править | править код]

Чрезвычайно ядовит. Арсин является мощнейшим ядом среди неорганических ядов. Среди соединений мышьяка наиболее токсичен. Он настолько ядовит, что сильнее некоторых органических ядов. По мощности сравним только со станнаном и стибином. ПДК 0,0003 мг/л. Оказывает кроверазрушающее действие. Канцерогенен: При длительном и частом воздействии на организм может вызвать злокачественные новообразования.
Применяют AsH3 для легирования полупроводниковых материалов мышьяком и для получения мышьяка высокой чистоты.
Синильная кислота — Википедия
| Синильная кислота | |||
|---|---|---|---|
 ( ({{{картинка малая}}}) | |||
| Традиционные названия | циановодород, синильная кислота | ||
| Хим. формула | CHN | ||
| Рац. формула | HCN | ||
| Состояние | бесцветный газ или бесцветная легколетучая жидкость | ||
| Молярная масса | 27,0253 г/моль | ||
| Плотность | 0,687 г/см³ | ||
| Динамическая вязкость | 0,201 Па·с | ||
| Энергия ионизации | 13,6 ± 0,1 эВ[1] | ||
| Температура | |||
| • плавления | −13,4 °C | ||
| • кипения | 26,7 °C | ||
| • вспышки | −17,8 °C | ||
| Пределы взрываемости | 5,6 ± 0,1 об.%[1] | ||
| Мол. теплоёмк. | (средняя для газа и жидкости) 1,97 Дж/(моль·К) | ||
| Давление пара | 630 ± 1 мм рт.ст.[1] | ||
| Константа диссоциации кислоты pKa{\displaystyle pK_{a}} | 9,21 | ||
| Растворимость | |||
| • в воде | в любых пропорциях | ||
| Показатель преломления | 1,2675 | ||
| Дипольный момент | 2,98 Д | ||
| Рег. номер CAS | 74-90-8 | ||
| PubChem | 768 | ||
| Рег. номер EINECS | 200-821-6 | ||
| SMILES | |||
| InChI | |||
| RTECS | MW6825000 | ||
| ChEBI | 18407 | ||
| Номер ООН | 1051 | ||
| ChemSpider | 748 и 19951400 | ||
| ЛД50 | 3,7 мг/кг (мыши, перорально) | ||
| Токсичность | Чрезвычайно токсична, СДЯВ | ||
| Пиктограммы ECB |   | ||
| NFPA 704 | | ||
| Приведены данные для стандартных условий (25 °C, 100 кПа), если не указано иное. | |||
 (
(Сини́льная (циа́нистоводородная) кислота́, цианистый водород, HCN[2] — бесцветная, очень летучая, легкоподвижная ядовитая жидкость, имеющая характерный запах горького миндаля[3].
Синильная кислота содержится в некоторых растениях, коксовом газе, табачном дыме, выделяется при термическом разложении нейлона, полиуретанов. Смешивается во всех соотношениях с водой, этанолом, диэтиловым эфиром.
Химические[править | править код]
Молекула HCN сильно полярна (μ = 0,96⋅10−29 Кл·м).
Безводный цианистый водород является сильно ионизирующим растворителем, растворённые в нём электролиты хорошо диссоциируют на ионы. Его относительная диэлектрическая проницаемость при 25 °C равна 107 (выше, чем у воды). Это обусловлено линейной ассоциацией полярных молекул HCN за счёт образования водородных связей.
Очень слабая одноосновная кислота К = 1,32⋅10−9 (18 °C). Образует с металлами соли — цианиды. Взаимодействует с оксидами и гидроксидами щелочных и щёлочноземельных металлов.
Пары синильной кислоты горят на воздухе фиолетовым пламенем с образованием Н2О, СО и N2. В смеси кислорода со фтором горит с выделением большого количества тепла:
- 2HCN+O2+F2→2HF+2CO+N2+1020{\displaystyle {\mathsf {2HCN+O_{2}+F_{2}\rightarrow 2HF+2CO+N_{2}+1020}}} кДж.
Синильная кислота широко применяется в органическом синтезе. Она реагирует с карбонильными соединениями, образуя циангидрины:
- RR′C=O+HCN→RR′C(OH)CN.{\displaystyle {\mathsf {RR’C\!=\!O+HCN\rightarrow RR’C(OH)CN}}.}
С хлором, бромом и иодом прямо образует циангалогениды:
- X2+HCN→XCN+HX.{\displaystyle {\mathsf {X_{2}+HCN\rightarrow XCN+HX}}.}
С галогеналканами — нитрилы (реакция Кольбе):
- RX+HCN→R−CN+HX.{\displaystyle {\mathsf {RX+HCN\rightarrow R\!-\!CN+HX}}.}
С алкенами и алкинами реагирует, присоединяясь к кратным связям:
- HCN+CH≡CH→Cu+Ch3=CHCN.{\displaystyle {\mathsf {HCN+CH\!\equiv \!CH{\xrightarrow {Cu^{+}}}CH_{2}\!=\!CHCN}}.}
- HCN+Ch3=Ch3 →Pd/Al2O3 Ch4Ch3CN.{\displaystyle {\mathsf {HCN+CH_{2}\!=\!CH_{2}\ {\xrightarrow {Pd/Al_{2}O_{3}}}\ CH_{3}CH_{2}CN}}.}
- HCN+RCH=NH→Cu+RCH(Nh3)CN.{\displaystyle {\mathsf {HCN+RCH\!=\!NH{\xrightarrow {Cu^{+}}}RCH(NH_{2})CN}}.}
Легко полимеризуется в присутствии основания (часто со взрывом). Образует аддукты, например, HCN-CuCl.
При разложении водой даёт формиат аммония, либо формамид
HCN+2h3O⟶HCOONh5{\displaystyle {\ce {HCN + 2h3O -> HCOONh5}}}
HCN+h3O⟶HCONh3{\displaystyle {\mathsf {HCN+H_{2}O\longrightarrow HCONH_{2}}}}
Физиологические[править | править код]
Синильная кислота является веществом, вызывающим кислородное голодание тканевого типа.[4] При этом наблюдается высокое содержание кислорода как в артериальной, так и в венозной крови и уменьшение таким образом артерио-венозной разницы, резкое понижение потребления кислорода тканями с уменьшением образования в них углекислоты. Синильная кислота и её соли, растворённые в крови, достигают тканей, где вступают во взаимодействие с трёхвалентной формой железа цитохромоксидазы. Соединившись с цианидом, цитохромоксидаза теряет способность переносить электроны на молекулярный кислород. Вследствие выхода из строя конечного звена окисления блокируется вся дыхательная цепь и развивается тканевая гипоксия. С артериальной кровью кислород доставляется к тканям в достаточном количестве, но не усваивается ими и переходит в неизмененном виде в венозное русло. Одновременно нарушаются процессы образования макроэргов, необходимых для нормальной деятельности различных органов и систем. Активизируется гликолиз, то есть обмен с аэробного перестраивается на анаэробный. Также подавляется активность и других ферментов — каталазы, пероксидазы, лактатдегидрогеназы.
Действие на нервную систему[править | править код]
В результате тканевой гипоксии, развивающейся под влиянием синильной кислоты, в первую очередь нарушаются функции центральной нервной системы.
Действие на дыхательную систему[править | править код]
В результате острого отравления наблюдается резкое увеличение частоты и глубины дыхания. Развивающуюся одышку следует рассматривать как компенсаторную реакцию организма на гипоксию. Стимулирующее действие синильной кислоты на дыхание обусловлено возбуждением хеморецепторов каротидного синуса и непосредственным действием яда на клетки дыхательного центра. Первоначальное возбуждение дыхания по мере развития интоксикации сменяется его угнетением вплоть до полной остановки. Причинами этих нарушений являются тканевая гипоксия и истощение энергетических ресурсов в клетках каротидного синуса и в центрах продолговатого мозга.
Действие на сердечно-сосудистую систему[править | править код]
Проникая в кровь, синильная кислота снижает способность клеток воспринимать кислород из притекающей крови. А так как нервные клетки больше остальных нуждаются в кислороде, они первыми страдают от её действия. В начальном периоде интоксикации наблюдается замедление сердечного ритма. Повышение артериального давления и увеличение минутного объёма сердца происходят за счёт возбуждения синильной кислотой хеморецепторов каротидного синуса и клеток сосудодвигательного центра, с одной стороны, и выброса катехоламинов из надпочечников и вследствие этого спазма сосудов — с другой. В дальнейшем артериальное давление падает, пульс учащается, развивается острая сердечно-сосудистая недостаточность и наступает остановка сердца.
Изменения в системе крови[править | править код]
Содержание в крови эритроцитов увеличивается, что объясняется рефлекторным сокращением селезёнки в ответ на развивающуюся гипоксию. Цвет венозной крови становится ярко-алым за счёт избыточного содержания кислорода, не поглощённого тканями. Артерио-венозная разница по кислороду резко уменьшается. При угнетении тканевого дыхания изменяется как газовый, так и биохимический состав крови. Содержание CO2 в крови снижается вследствие меньшего образования и усиленного его выделения при гипервентиляции. Это приводит в начале развития интоксикации к газовому алкалозу, который меняется метаболическим ацидозом, что является следствием активации процессов гликолиза. В крови накапливаются недоокисленные продукты обмена. Увеличивается содержание молочной кислоты, нарастает содержание ацетоновых тел, отмечается гипергликемия. Нарушение окислительно-восстановительных процессов в тканях приводит к гипотермии. Таким образом, синильная кислота и её соли вызывают явления тканевой гипоксии и связанные с ней нарушения дыхания, кровообращения, обмена веществ, функции центральной нервной системы, выраженность которых зависит от тяжести интоксикации.
Показано, что нейроны способны вырабатывать эндогенную синильную кислоту (цианистый водород, HCN) после их активации эндогенными или экзогенными опиоидами и что образование нейронами эндогенной синильной кислоты повышает активность NMDA-рецепторов и, таким образом, может играть важную роль в передаче сигнала между нейронами (нейротрансмиссии). Более того, образование эндогенного цианида оказалось необходимым для проявления в полном объёме анальгетического действия эндогенных и экзогенных опиоидов, а вещества, снижающие образование свободной HCN, оказались способны уменьшать (но не полностью устранять) анальгетическое действие эндогенных и экзогенных опиоидов. Выдвинуто предположение, что эндогенная синильная кислота может являться нейромодулятором[5].
Известно также, что стимуляция мускариновых холинорецепторов клеток феохромоцитомы в культуре повышает образование ими эндогенной синильной кислоты, однако стимуляция мускариновых холинорецепторов ЦНС в живом организме крысы приводит, наоборот, к снижению образования эндогенной синильной кислоты[6].
Также показано, что синильная кислота выделяется лейкоцитами в процессе фагоцитоза и способна убивать патогенные микроорганизмы[5].
Возможно, что вазодилатация, вызываемая нитропруссидом натрия, связана не только с образованием окиси азота (механизм, общий для действия всех сосудорасширяющих препаратов группы нитратов, таких как нитроглицерин, нитросорбид), но и с образованием цианида. Возможно, что эндогенный цианид и образующийся при его обезвреживании в организме тиоцианат играют роль в регуляции функций сердечно-сосудистой системы, в обеспечении вазодилатации и являются одними из эндогенных антигипертензивных веществ[7].
В настоящий момент существуют три наиболее распространённых метода получения синильной кислоты в промышленных масштабах:
2Nh4+2Ch5+3O2→Pt2HCN+6h3O.{\displaystyle {\mathsf {2NH_{3}+2CH_{4}+3O_{2}{\xrightarrow {Pt}}2HCN+6H_{2}O}}.}
- Метод BMA (Blausäure aus Methan und Ammoniak), запатентованный фирмой Degussa: прямой синтез из аммиака и метана без воздуха в присутствии платинового катализатора при высокой температуре:
Nh4+Ch5→PtHCN+3h3.{\displaystyle {\mathsf {NH_{3}+CH_{4}{\xrightarrow {Pt}}HCN+3H_{2}}}.}
KCN+h3O+CO2⟶HCN+KHCO3{\displaystyle {\mathsf {KCN+H_{2}O+CO_{2}\longrightarrow HCN+KHCO_{3}}}}
2h4[Fe(CN)6] →T Fe[Fe(CN)6]+6HCN{\displaystyle {\mathsf {2H_{3}[Fe(CN)_{6}]\ {\xrightarrow {T}}\ Fe[Fe(CN)_{6}]+6HCN}}}
3h5[Fe(CN)6] →100oC Fe2[Fe(CN)6]+12HCN{\displaystyle {\mathsf {3H_{4}[Fe(CN)_{6}]\ {\xrightarrow {100^{o}C}}\ Fe_{2}[Fe(CN)_{6}]+12HCN}}}(в присутствии влаги)
HCl+NaCN⟶HCN+NaCl{\displaystyle {\ce {HCl + NaCN->HCN + NaCl}}}
H++NaCN⟶HCN+Na+{\displaystyle {\ce {H+ + NaCN ->HCN + Na+}}}
Эта реакция иногда является основой случайных отравлений, потому что кислота превращает нелетучую цианидную соль в газообразный циановодород.
- Реакцией монооксида углерода с аммиаком:
Nh4+CO→ThO2HCN+h3O.{\displaystyle {\mathsf {NH_{3}+CO{\xrightarrow {ThO2}}HCN+H_{2}O}}.}
- Фотолиз метана в бескислородной атмосфере:
2Ch5+N2⟶2HCN+3h3{\textstyle {\mathsf {2CH_{4}+N_{2}\longrightarrow 2HCN+3H_{2}}}}
В химическом производстве[править | править код]
Является сырьём для получения акрилонитрила, метилметакрилата, адипонитрила и других соединений. Синильная большое число её производных используются при извлечении благородных металлов из руд, при гальванопластическом золочении и серебрении, в производстве ароматических веществ, химических волокон, пластмасс, каучука, органического стекла, стимуляторов роста растений, гербицидов.
Как отравляющее веществo[править | править код]
Впервые в роли боевого отравляющего вещества синильная кислота была использована французской армией 1 июля 1916 года на реке Сомме[9]. Однако из-за отсутствия кумулятивных свойств и малой стойкости на местности её последующее использование в этом качестве прекратилось.
Синильная кислота являлась основной составляющей препарата «Циклон Б», который был наиболее популярным в Европе во время Второй мировой войны инсектицидом, а также использовался нацистами для убийства людей в концентрационных лагерях. В некоторых штатах США синильная кислота использовалась в газовых камерах в качестве отравляющего вещества при исполнении приговоров смертной казни; в последний раз это было сделано в Аризоне в 1999 году[10]. Смерть, как правило, наступает в течение 5—15 минут.
Соли синильной кислоты называются цианидами. Все цианиды, как и сама кислота, очень ядовиты. Цианиды подвержены сильному гидролизу. При хранении водных растворов цианидов при доступе диоксида углерода они разлагаются:
- KCN+h3O+CO2⟶HCN+KHCO3{\displaystyle {\mathsf {KCN+H_{2}O+CO_{2}\longrightarrow HCN+KHCO_{3}}}}
- KCN+2h3O⟶Nh4+HCOOK{\displaystyle {\mathsf {KCN+2H_{2}O\longrightarrow NH_{3}+HCOOK}}}
Ион CN− (изоэлектронный молекуле СО) входит как лиганд в большое число комплексных соединений d-элементов. Комплексные цианиды в растворах очень стабильны.
Цианиды тяжёлых металлов термически неустойчивы; в воде, кроме цианида ртути (Hg(CN)2), нерастворимы. При окислении цианиды образуют соли — цианаты:
- 2KCN+O2⟶2KOCN{\displaystyle {\mathsf {2KCN+O_{2}\longrightarrow 2KOCN}}}
Многие металлы при действии избытка цианида калия или цианида натрия дают комплексные соединения, что используется, например, для извлечения золота и серебра из руд:
- 8NaCN+4Au+O2+2h3O⟶4Na[Au(CN)2]+4NaOH{\displaystyle {\mathsf {8NaCN+4Au+O_{2}+2H_{2}O\longrightarrow 4Na[Au(CN)_{2}]+4NaOH}}}
Токсичность и биологические свойства[править | править код]

Синильная кислота — сильнейший яд общетоксического действия, блокирует клеточную цитохромоксидазу, в результате чего возникает выраженная тканевая гипоксия. Половинные летальные дозы (LD50) и концентрации для синильной кислоты[11]:
- Мыши:
- перорально (ORL-MUS LD50) — 3,7 мг/кг;
- при вдыхании (IHL-MUS LC50) — 323 м.д.;
- внутривенно (IVN-MUS LD50) — 1 мг/кг.
- Кролики, внутривенно (IVN-RBT LD50) < 1 мг/кг;
- Человек, минимальная опубликованная смертельная доза перорально (ORL-MAN LDLo) < 1 мг/кг.
При вдыхании синильной кислоты в небольших концентрациях наблюдается царапанье в горле, горький вкус во рту, головная боль, тошнота, рвота, боли за грудиной. При нарастании интоксикации уменьшается частота пульса, усиливается одышка, развиваются судороги, наступает потеря сознания. При этом цианоз отсутствует (содержание кислорода в крови достаточное, нарушена его утилизация в тканях).
При вдыхании синильной кислоты в высоких концентрациях или при попадании её внутрь появляются клонико-тонические судороги и почти мгновенная потеря сознания вследствие паралича дыхательного центра. Смерть может наступить в течение нескольких минут.
В организме человека метаболитом синильной кислоты является роданид (тиоцианат) SCN−, образующийся при её взаимодействии с серой под действием фермента роданазы.
Для лечения отравлений синильной кислотой известно несколько антидотов, которые могут быть разделены на две группы. Лечебное действие одной группы антидотов основано на их взаимодействии с синильной кислотой с образованием нетоксичных продуктов. К таким препаратам относятся, например, коллоидная сера и различные политионаты, переводящие синильную кислоту в малотоксичную роданистоводородную кислоту, а также альдегиды и кетоны (глюкоза, диоксиацетон и др.), которые химически связывают синильную кислоту с образованием циангидринов. К другой группе антидотов относятся препараты, вызывающие образование в крови метгемоглобина: синильная кислота связывается метгемоглобином и не доходит до цитохромоксидазы. В качестве метгемоглобинообразователей применяют метиленовую синь, а также соли и эфиры азотистой кислоты.
Сравнительная оценка антидотных средств: метиленовая синь предохраняет от двух смертельных доз, тиосульфат натрия и тетратиосульфат натрия — от трёх доз, нитрит натрия и этилнитрит — от четырёх доз, метиленовая синь совместно с тетратиосульфатом — от шести доз, амилнитрит совместно с тиосульфатом— от десяти доз, азотистокислый натрий совместно с тиосульфатом — от двадцати смертельных доз синильной кислоты.
ПДК[12] в воздухе рабочей зоны равна 0,3 мг/м3 (максимально-разовая). По данным[13] при опасной концентрации люди скорее всего не почувствуют запаха; а согласно[14] порог восприятия запаха может быть 5,6 мг/м3. Поэтому использование широко распространённых фильтрующих СИЗОД в сочетании с «заменой фильтров по появлении запаха под маской» (как это почти всегда рекомендуется в РФ поставщиками СИЗОД) приведёт к чрезмерному воздействию синильной кислоты на, по крайней мере, часть работников
Дихромат натрия — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 21 декабря 2017; проверки требуют 14 правок. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 21 декабря 2017; проверки требуют 14 правок.Дихрома́т на́трия (бихромат натрия, натриевый хромпик) — неорганическое химическое соединение, натриевая соль дихромовой кислоты. Существует дигидрат дихромата натрия (Na2Cr2O7·2H2O) и безводная соль.
Полупродукт в производстве хрома, практически вся хромовая руда перерабатывается в дихромат натрия.
Следует учитывать, что дихромат натрия очень токсичен.
Химические[править | править код]
С точки зрения реакционной способности и внешнего вида дихромат натрия и дихромат калия очень похожи. Соль натрия, однако, в двадцать раз лучше растворяется в воде, чем соль калия (49 г/л при 0 °C) и её эквивалентная масса также меньше — поэтому она является наиболее часто используемым веществом[источник не указан 503 дня].
Схожестью хромовой кислоты и дихромата натрия является их общее свойство — они являются сильными окислителями. По отношению к калийной соли, основным преимуществом дихромата натрия является его большая растворимость в воде и в полярных растворителях, например таких как уксусная кислота.
В области органического синтеза это соединение окисляет бензилы и аллильную группу C—H соединений до карбонильных производных. Так, например, 2,4,6-тринитротолуол окисляется до соответствующих карбоновых солей. Кроме того, 2,3-диметилнафталин (англ. 2,3-dimethylnaphthalene) окисляется в присутствии Na2Cr2O7 до 2,3-нафтилдикарбоновой кислоты (англ. 2,3-naphthalenedicarboxylic acid).
Дихромат натрия образуется в больших масштабах из руд, содержащих оксид хрома(III).
Сначала руду сплавляют, как правило, с карбонатом натрия при температуре около 1000 °C в присутствии воздуха (источник кислорода):
- 2Cr2O3+4Na2CO3+3O2⟶4Na2CrO4+4CO2{\displaystyle {\ce {2 Cr2O3 + 4 Na2CO3 + 3 O2 -> 4 Na2CrO4 + 4 CO2}}}
На данном этапе другие компоненты руды, такие как алюминий и железо, плохо растворимы. Окисление в результате реакции водного экстракта серной кислоты или углекислого газа даёт дихромат натрия, который выделяется как дигидрат при кристаллизации. Соединения хромаVI являются токсичными, в частности, при получении в виде пыли производящие его заводы могут быть подвержены строгим правилам. Например, чтобы снизить его токсичность, его сливают в сточные воды, где происходит восстановление с получением хромаIII, который является менее опасным для окружающей среды.
Используют при дублении кож и в электрических элементах, как компонент биозащитных составов для древесины.
Как и все соединения шестивалентного хрома, дихромат натрия очень токсичен. Кроме того, он известный канцероген. Вещество очень токсично для водных организмов, может вызвать долговременные изменения в водной экосистеме.