3.2 Кислотно-основное равновесие
3.2.1 Типы протолитических реакций
1. Реакции протолиза (ионизации).
К ним относятся реакции взаимодействия кислоты или основания с водой:

К-та 1 осн.2 к-та 2 осн.1
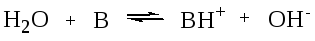
К-та 1 осн.2 к-та 2 осн. 1
2. Реакции автопротолиза, связанные с передачей протона от одной молекулы воды к другой.

Реакции гидролиза
CH3COO— + H2O ←→ CH3COOH + OH—
осн.2 к-та 1 к-та 2 осн.1
Кислотно-основные реакции
NH3 + HCl → NH4+ + Cl—
осн.2 к-та 1 к-та 2 осн.1
С точки зрения аналитики выделяют следующие типы реакций:
1) с переносом протона – кислотно-основные;
2) с переносом электрона – ОВ реакции;
3) с переносом электронных пар с образованием связей по донорно-акцепторному механизму – реакции комплексообразования.
2.2.2 Константа кислотности и основности. Расчеты рН
Способность кислоты отдавать протон, а основания принимать его (т.е. силу кислот и оснований) можно охарактеризовать константами равновесий,
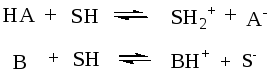 HS
– растворитель
HS
– растворитель
которые называют константами кислотности (Ка) и основности (Кb).

Активность растворителя – величина постоянная (табличные данные)

Положения кислотно-основных равновесий

и величины соответствующих констант кислотности и основности зависят от природы растворителя.
Если растворитель – более сильный акцептор протонов, чем вода (например, аммиак), то в нем сила кислот возрастает. Так кислоты слабые в водных растворах могут быть сильными в аммиаке.
Чем сильнее основные свойства растворителя, тем больше кислот нивелируется в нем.
Аналогично, чем сильнее кислотные свойства растворителя, тем больше оснований нивелируется в нем.
При переходе от более к менее основному растворителю сильные кислоты могут быть слабыми (напр., HCl и HClO4 в воде – сильные кислоты , а в ледяной уксусной кислоте становятся слабыми).
Расчет рН
Расчеты кислотно-основных равновесий используют для:
1) нахождения рН раствора по известным равновесным концентрациям;
2) определения равновесных концентраций по известному значению рН
рН – важная оценка для биологических жидкостей.
Для живых организмов характерно поддержание кислотно-основного состояния на определенном уровне. Это находит выражение в достаточно постоянных значениях рН биологических сред и способности восстанавливать нормальные значения рН при воздействии протолитов.
Система, поддерживающая протолитический гомеостаз, включает в себя не только физиологические механизмы (легочную и почечную компенсации), но и физико-химическое действие, ионный обмен, диффузию.
В аналитической химии важно знать концентрации всех частиц в растворе кислоты или основания после установления равновесия, в частности концентрацию ионов Н+ (рН).
 —
слабый электролит
—
слабый электролит
— сильный электролит
Чистая вода

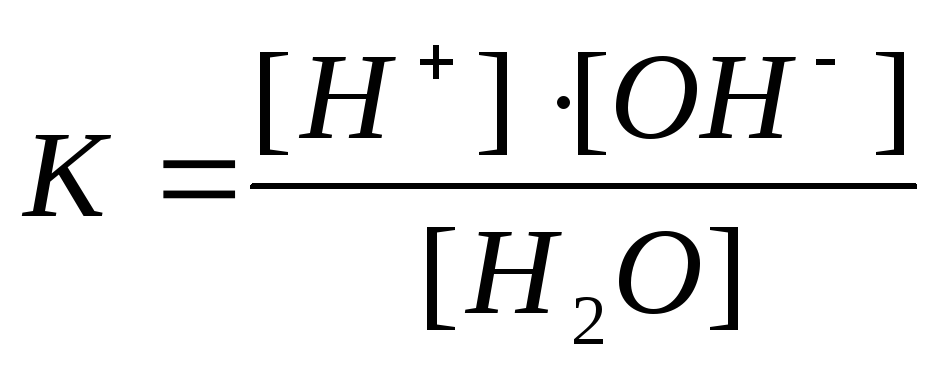
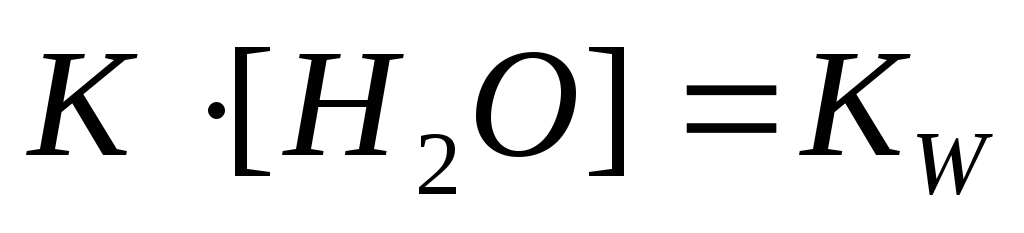
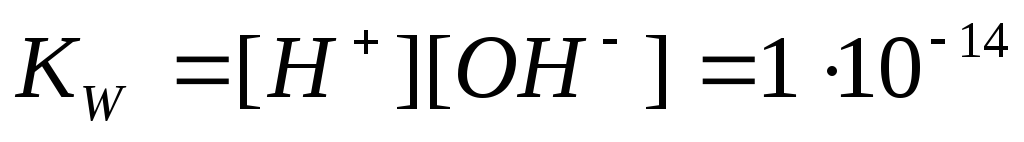



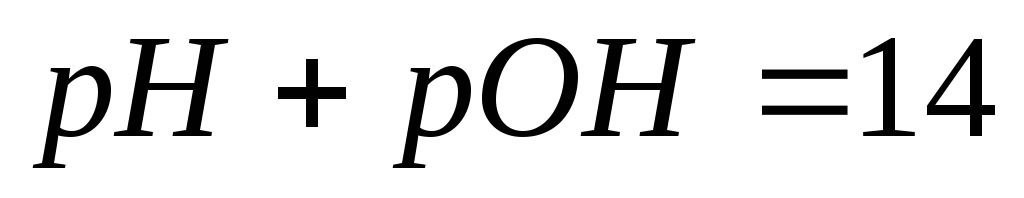
Чистой воды не существует. Морская вода содержит почти все химические элементы.
Растворы слабых кислот
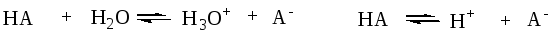
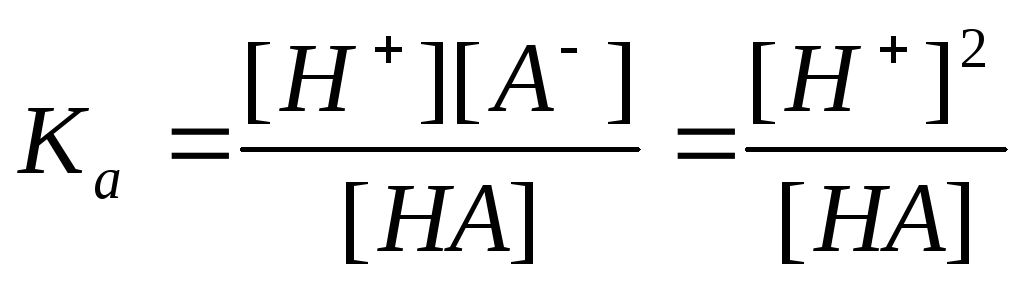 Т.к.
Т.к. 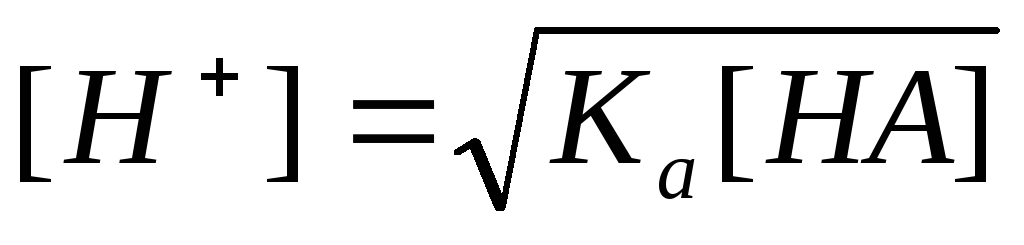 ,
то
,
то 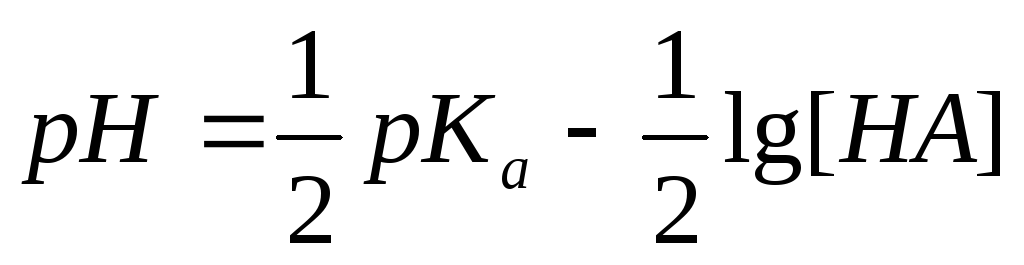
Пример 1. Рассчитать рН 0,1М раствора уксусной кислоты.
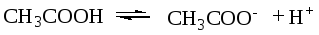
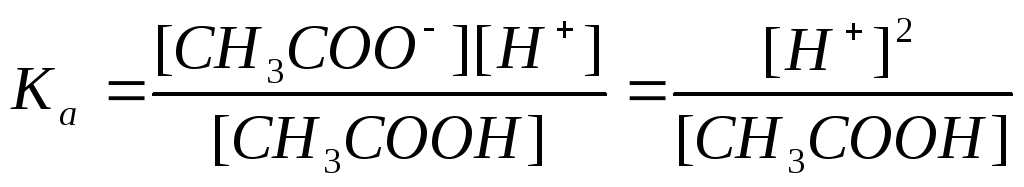
 М
М
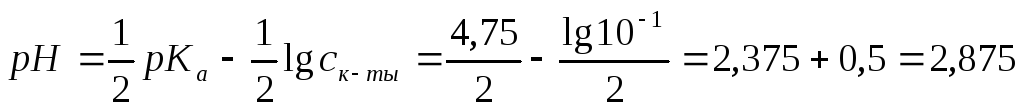 или
или
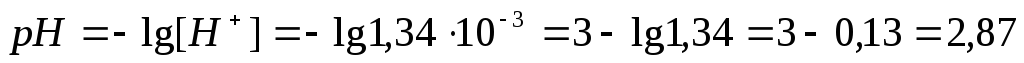
Растворы слабых оснований

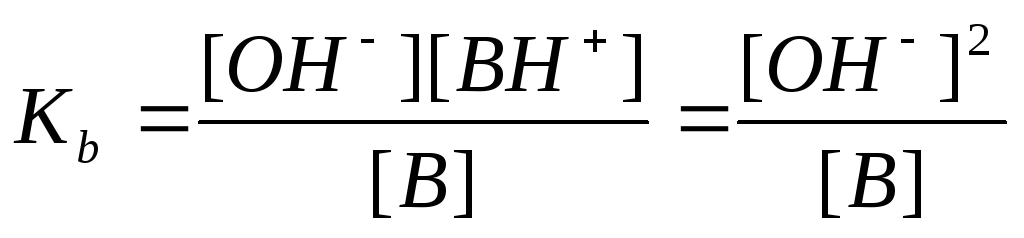
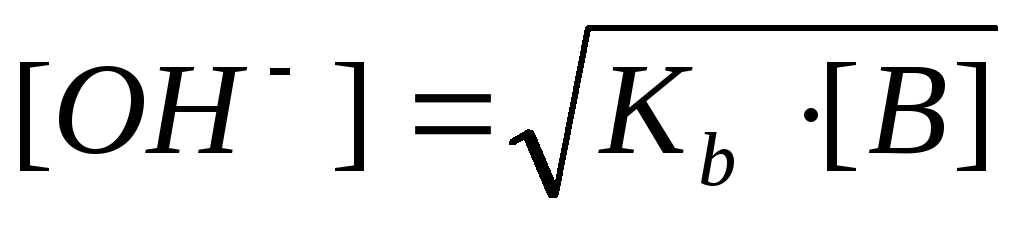
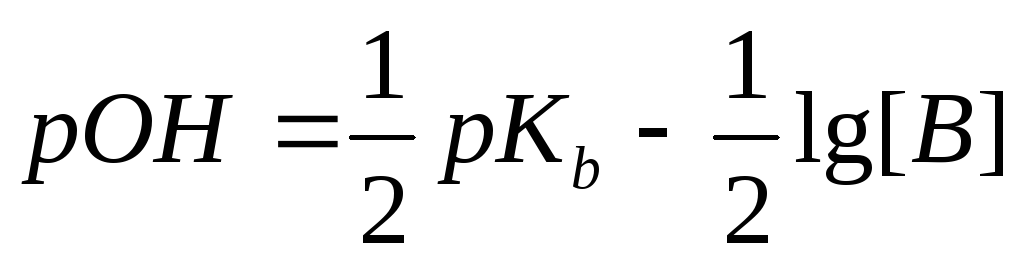
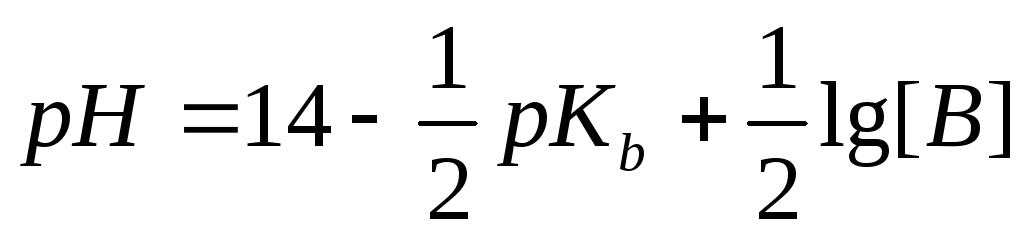
Пример 2. Рассчитать рН 0,1 М раствора аммония гидроксида.
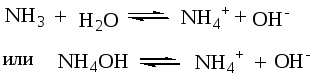



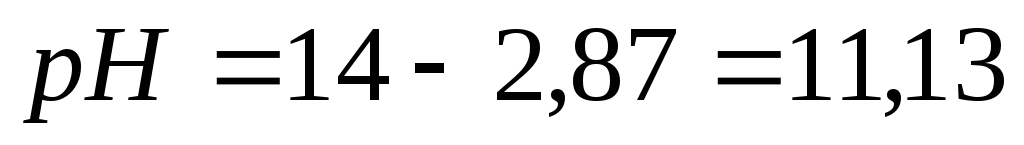
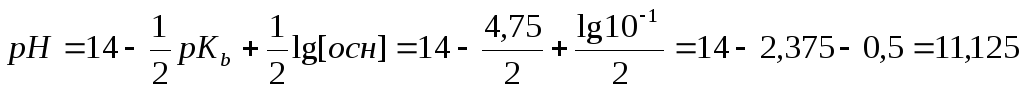
Растворы сильных кислот

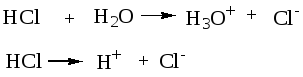
Для учета влияния электростатического взаимодействия ионов введено понятие ионной силы раствора. Она зависит от концентрации иона и его заряда.
Для сильных электролитов закон действия масс выполняется, если пользуются активностями. Активность учитывает концентрацию реагентов, меж-ионное взаимодействие (ион-ионное, ион-дипольное, диполь-дипольное, водородные связи).
Согласно теории Дебая и Хюккеля
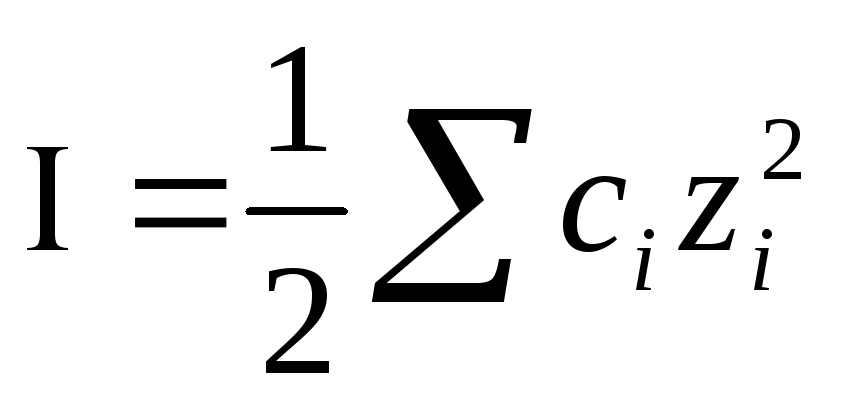

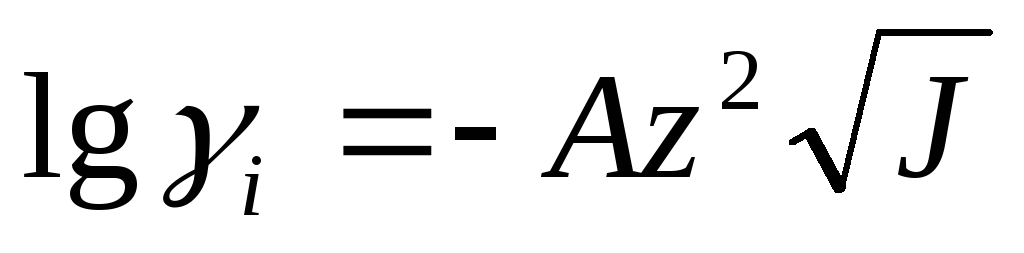 — зависимость
коэффициента подвижности от ионной
силы
— зависимость
коэффициента подвижности от ионной
силы
А зависит от диэлектрической постоянной растворителя и температуры системы. При t=25°С А=0,512 и для бинарного электролита
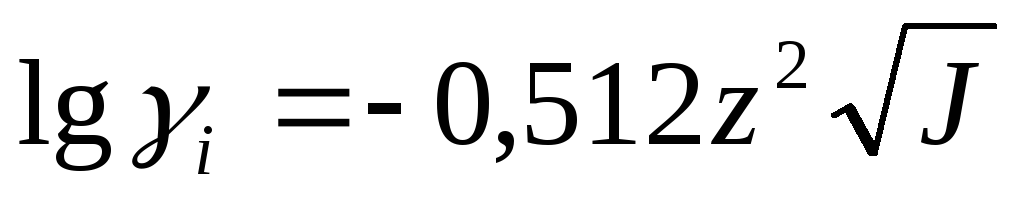
Пример3. Рассчитать рН 0,1М р-ра НСl.
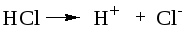
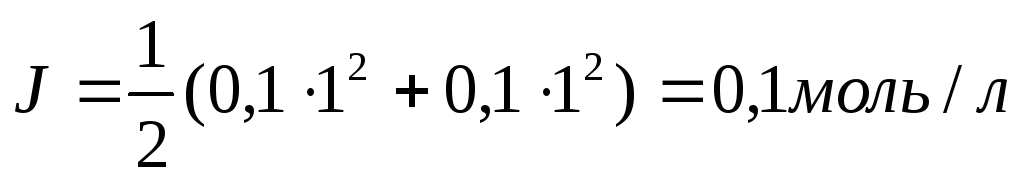

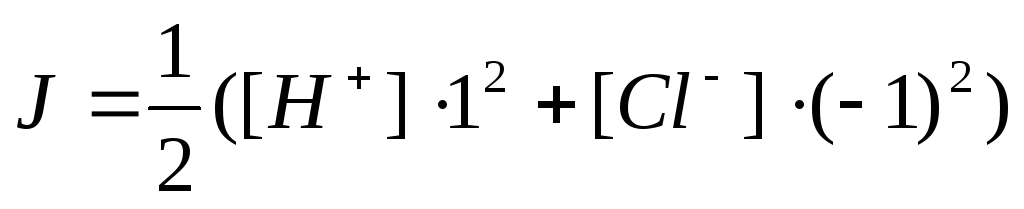
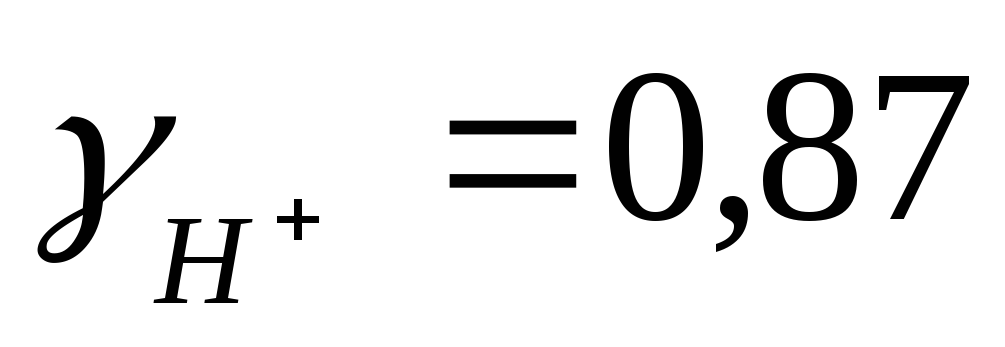
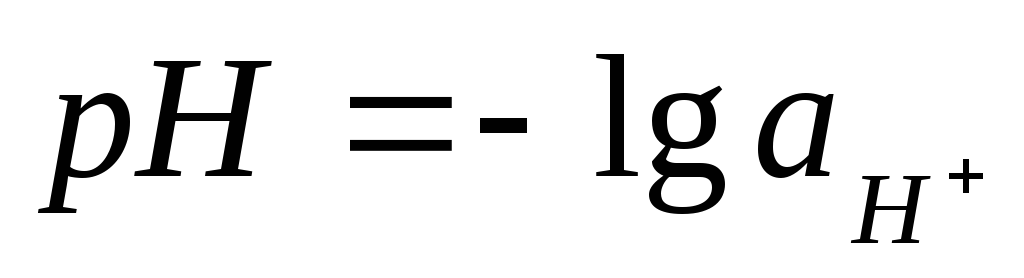
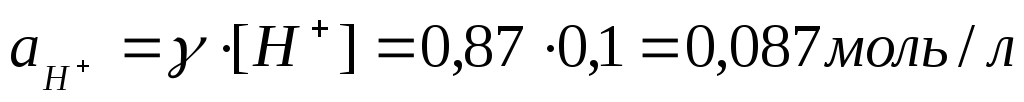
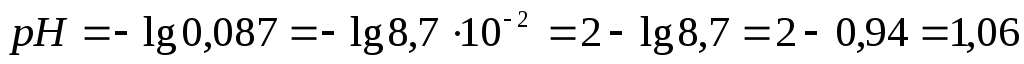
Растворы сильных оснований
Пример 4. Рассчитать рН 0,1 н. раствора натрия гидроксида.

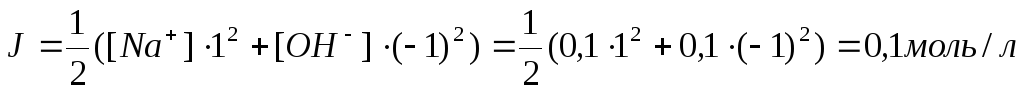

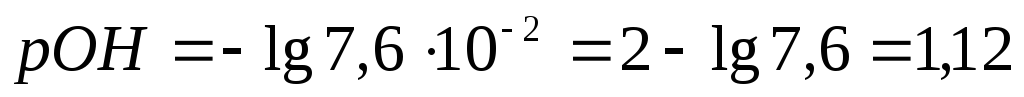
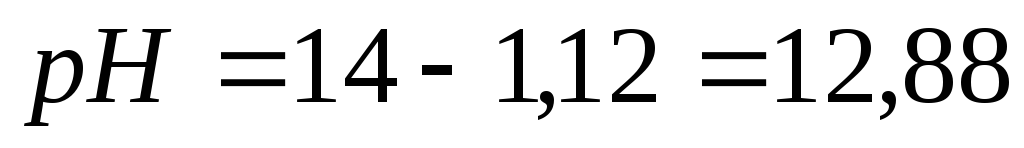
3.3 Протолитическое равновесие в буферных растворах
В широком смысле буферными называют системы, поддерживающие определенное значение какого-либо параметра при изменении состава.
Буферные растворы могут быть кислотно-основными – поддерживают постоянное значение рН при введении кислот или оснований; окислительно-восстановительными – сохраняют постоянным потенциал системы при введении окислителей или восстановителей; известны металлобуферные растворы.
Буферный раствор представляет собой сопряженную пару; в частности, кислотно-основной буфер – сопряженную кислотно-основную пару:
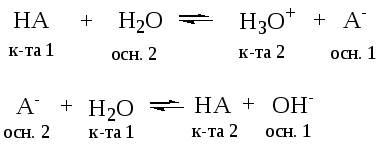
Кислотность и основность органических соединений
15
Цель занятия:
Сформировать у студентов представление о влиянии кислотно-основных свойств органических соединений на многие физико-химические и биологические процессы, протекающие в условиях организма.
Научить студентов определять кислотно-основные свойства спиртов, фенолов, тиолов, и аминов в зависимости от их строения.
Студент должен знать:типы кислот и оснований Бренстеда.
Студент должен уметь: определять кислотные и основные свойства органических соединений.
Современные представления о кислотах и основаниях. Бренстеда и Лоури
Важными аспектами реакционной способности органических соединений является их кислотные и основные свойства. Для описания кислотных и основных свойств химических соединений существует несколько теорий – теория Бренстеда и Лоури, теория Льюиса и ряд других. Наиболее распространенной является теория Бренстеда и Лоури, или протолитическая теория.
По теории Бренстеда – Лоури кислоты – это нейтральные молекулы или ионы, способные отдавать протон (доноры протона), а основания — это нейтральные молекулы или ионы, способные присоединять протон (акцепторы протона).
По теории Льюиса кислоты – это нейтральные молекулы или ионы, способные присоединять электронную пару (акцепторы электронной пары), а основания – это нейтральные молекулы или ионы, способные отдавать электронную пару (доноры электронной пары).
Из этого следует, что теоретически любое соединение, в состав которого входит атом водорода может его отдавать в виде протона и, проявлять свойства кислоты. Способность отдавать протон могут проявлять не только нейтральные молекулы, но заряженные частицы – катионы (NH4+) и анионы кислот, например HCl, ROH, HSO4— и др.
В роли оснований могут выступать анионы – частицы, несущие отрицательный заряд, например С1—, OH—, HSO4, NH3. Основаниями могут быть и нейтральные молекулы, в состав которых входит гетероатом, например, азот, сера, кислород, содержащие неподелённую пару электронов, например спирты ROH.
Нейтральные молекулы или заряженные ионы, способные в зависимости от природы второго компонента проявлять свойства кислот или оснований называются амфотерными.
Теория Бренстеда – Лоури. Сопряженные кислоты и основания.
Кислоты и основания проявляют свои свойства только в присутствие друг друга, Ни одно вещесвто не будет отдавать протон, если в системе нет акцептора протона – основания, и наоборот.т.е. они образуют сопряжённую кислотно-основную пару в которой чем сильнее кислота, тем слабее сопряженное ей основание, и чем сильнее основание, тем слабее сопряженная ему кислота.
Кислота, отдавая протон, превращается в сопряженное основание, а основание приняв протон, превращается в сопряженную кислоту. Кислоту обычно обозначают АН, а основание – В
Например: НС1↔ Н+ + С1—, НС1 – сильная кислота; С1— ион – сопряженное слабое основание;
СН3СООН ↔ СН3СОО— + Н+, СН3СООН – слабая кислота, а СН3СОО— — ион сопряженное сильное основание.
Общий вид можно представить так: Н+│ : А + В ↔ Н:В+ + А:—
к-та основ сопр. сопр.
к-та основ-е
Мы уже сказали, что кислотные свойства соединений обнаруживаются только в присутствие основания, а основные свойства – в присутствие кислоты, т.е. в соединениях существует определённое кислотно — основное равновесие, для изучения которого в качестве растворителя используется Н2О. По отношению к Н2О как к кислоте или как основанию определяют кислотно-основные свойства соединений.
Для слабых электролитов кислотность количественно оценивается Крав реакции, которая заключается в переносе Н+ от кислоты к Н2О как основанию.
СН3СООН + Н2О ↔ СН3СОО— + Н3О+
к-та основ-е основание кислота
СН3СОО— — ацетат ион, сопряженное основание;
Н3О+ — ион гидроксония, сопряженная кислота.
Используя значение константы равновесия этой реакции и учитывая, что концентрация Н2О практически постоянна, можно определить произведение К ·[H2O] называемое константой кислотности К кислотности (Ка).
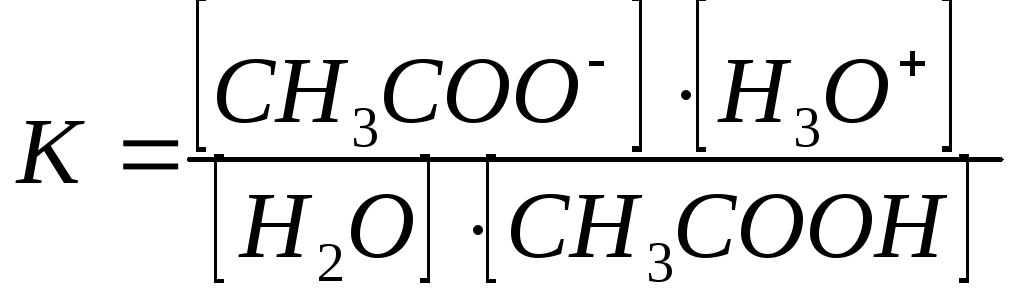 ;
;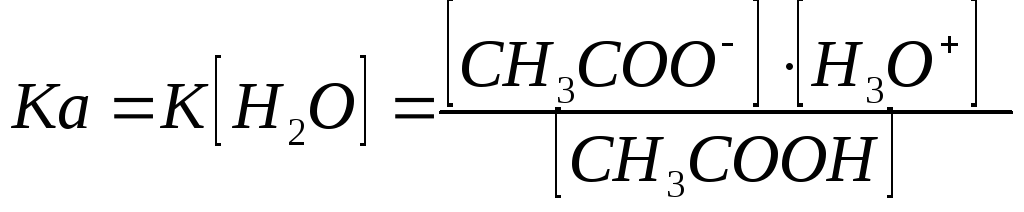
Чем больше Ка, тем сильнее кислота. Для СН3СООН Ка = 1,75 · 10-5. такие малые величины неудобны в практической работе, поэтому Ка выражают через рКа (рК = -ℓg Ка). Для СН3СООН рКа = 4,75. Чем меньше величина рКа, тем сильнее кислота.
Сила оснований определяется величиной рКВН+.
Количественная характеристика кислот и оснований по теории Бренстеда – Лоури.
Л6. ПРОТОЛИТИЧЕСКИЕ РАВНОВЕСИЯ И ПРОЦЕССЫ.
Согласно теории электролитической диссоциации Аррениуса кислотой является любое вещество в состав, которого входит ион водорода, основанием – гидроксид ион. Но в органической и биологической химии для оценки кислотности и основности используютпротолитическую теорию кислот и оснований Бренстеда – Лоури (1923г.).По теории понятия кислотность и основность связаны с переносом протона.
Кислотойявляется любая частица, способная отдать ион водорода.
NH4+ → H+ + NH30 HCl → H+ + Cl—
Основаниемявляется любая частица, способная принять ион водорода.
H2O + H+ → H3O+ NO3— + H+→ HNO3
По теории кислота и основание должны находиться в сопряженной системе, т.е. кислота не может существовать без основания.
HA + B = A— + BH+
кислота + основание = сопряженное основание + сопряженная кислота
Из этого уравнения следует: кислота и основание образуют кислотно-основные пары. Чем сильнее кислота, тем слабее будет сопряженное с ней основание и наоборот.
Пример 1:..
СН3СООН + НОН ↔ СН3СОО—+ Н3О+
кислота основание сопряж. основание сопряж. кислота
Поскольку этот процесс обратим, то он характеризуется равновесием:
Кс= [СН3СОО—]·[Н3О+] / [СН3СООН]·[НОН]
Кс· [Н2О] = [СН3СОО—]·[Н3О+] / [СН3СООН]
Ка= [СН3СОО—]·[Н+] / [СН3СООН] –константа кислотности
Чем больше константа кислотности, тем сильнее кислота, например: Ка (СН3СООН) = 1,75·10-5; Ка (Н2СО3) = 4,7·10-7.
Так как величины констант кислотности очень малы, то удобнее использовать не абсолютные значения констант кислотности, а их отрицательные логарифмы – показатели кислотности — рКа.
рКа= -lgКа
рКа(СН3СООН) = -lg1,75·10-5= 4,76
рКа(Н2СО3) = -lg4,7·10-7= 6,33
Чем больше показатель кислотности, тем слабее кислота и наоборот.
Пример 2:..
NH3+HOH↔NH4++OH—
основание кислота сопряж. кислота сопряж. основание
Кс= [NH4+]·[ОH—] / [NН3]·[НОН]
Кс· [НОН] = [NH4+]·[ОH—] / [NН3]
Кв= [NH4+]·[ОH—] / [NН3] –константа основности.
Чем выше константа основности, тем сильнее основание. Так как величины констант основности очень малы, то удобнее использовать не абсолютные значения констант основности, а их отрицательные логарифмы – показатели основности – рКв.
рКв= -lgКв
Чем меньше показатель основности, тем сильнее основание и наоборот.
Диссоциация воды. Ионное произведение воды.
..
НОН + НОН = ОН—+ Н3О+
кислота основание сопряж. основание сопряж. кислота
Вода по данной теории может быть как кислотой, так и основанием. Если процесс равновесный, то мы можем охарактеризовать его константой диссоциации – Кд.
Кд= [ОН—]·[Н3О+] / [НОН]·[НОН] = [ОН—]·[Н+] / [НОН]
Кд · [НОН] = [ОН—]·[Н+]
Кw= [ОН—]·[Н+] = 1·10-14 моль/л –ионное произведение воды.
В воде [H+] = [ОН—] = 1·10-7 моль/л; 10-1÷ 10-5– кислая среда, 10-5÷ 10-7– слабокислая среда, 10-7– нейтральная среда, 10-7÷ 10-9– слабощелочная среда, 10-9÷ 10-14– щелочная среда.
На практике чаще пользуются не абсолютным значением ионного произведения воды, а отрицательным логарифмом равновесной концентрации ионов водорода или гидроксид ионов.
рН = -lg[H+]; рОН = -lg[ОН—]
рН + рОН = 14
Для водных растворов рН = 1 ÷ 5 – кислая среда; рН = 5 ÷ 7 – слабокислая среда; рН = 7 – нейтральная среда; рН = 7 ÷ 9 – слабощелочная среда; рН = 9 ÷ 14 – щелочная среда.
Показатель кислотности для слабых кислот и оснований высчитывается по формулам:
рНслабой кислоты = ½ рКа– ½lgС(fэ)кислоты
рНслабого основания = 14 – ½ рКв+ ½lgС(fэ)основание
Внутренняя среда организма характеризуется постоянством концентрации ионов водорода. Это явление называют изогидрия. Например: рН крови = 7,1 ÷ 7,2; желудочного сока = 0,9 ÷ 1,1; пузырной желчи = 5,4 ÷ 6,9; грудного молока = 6,6 ÷ 6,8; слезной жидкости = 7,4. Изменения рН приводит к нарушению деятельности ферментов, регуляции осмотического давления и может приводить даже к гибели клеток. Сдвиг рН в кислую сторону, называетсяацидоз, а в щелочную –алкалоз.Так при сахарном диабете (кетоацидоз) наблюдается ацидоз, а при потери кислоты в случае неукротимой рвоты — алкалоз. Постоянство рН внутренних сред организма поддерживается работой почек, легких, печени – физиологический механизм, но основную роль играет химический механизм – за счет буферных систем.
Кислотность и щелочность почвы
Всякая почва обладает определенной реакцией, которая проявляется при взаимодействии с водой или растворами солей и может быть кислой, щелочной и нейтральной.
Кислотность почвы. Почвы, не насыщенные основаниями, обладают кислотностью, которая вызывается ионами водорода. В зависимости от того, в каком состоянии они находятся в почве, кислотность может быть активной, или актуальной, и потенциальной.
Под активной кислотностью понимают концентрацию свободных водородных ионов в почвенном растворе.
Источниками свободного водорода в почвенном растворе могут быть растворимые органические кислоты, образующиеся после разложения органических остатков, и углекислота, возникающая при растворении углекислого газа в воде. Углекислота, диссоциируя на Н+ и HCOj», подкисляет почвенный раствор. Активная кислотность частично вызывается и десорбцией обменных ионов водорода поглощающим комплексом.
Факторами активной кислотности в почве могут быть и некоторые минеральные соли А1 и Fe. Известно, что соли слабых оснований и сильных кислот в водных растворах гидролитически расщепляются, освобождая кислоту. Примером может служить хлористый алюминий, который при взаимодействии с водой расщепляется следующим образом:
А1С13 + ЗНаО = А1 (ОН)3 + ЗНС1.
Образующаяся при этом соляная кислота создает кислую реакцию раствора.
Эти явления наблюдаются только в почвах, не насыщенных основаниями. В почвах, насыщенных основаниями, активной кислотности нет.
Активная кислотность определяется в лабораториях в водной вытяжке из почвы. Количественно она выражается символом рН, который представляет собой отрицательный десятичный логарифм концентрации водородных ионов в почвенном растворе. При кислой реакции раствора рН меньше 7, при нейтральной равен 7, а при щелочной больше 7.
По величине рН почвы делят на следующие группы:
Сильнокислые 3—4
Нейтральные 7
Кислые 4—5,5
Щелочные 7—8
Слабокислые 5,5—6,5
Сильнощелочные 8—9
Кислую реакцию имеют подзолистые, дерново-подзолистые и болотные почвы, нейтральную — главным образом черноземы, щелочную — каштановые почвы, сероземы и солонцы.
Потенциальная кислотность обусловлена ионами водорода и алюминия, находящимися в поглощенном состоянии.
Кроме поглощенного водорода, обменную кислотность в почве создают обменнопоглощенные ионы алюминия, которые при взаимодействии с растворами нейтральных солей могут переходить в раствор.
Обменная кислотность наиболее ярко выражена в подзолистых почвах и красноземах. В почвах, имеющих слабокислую, нейтральную и щелочную реакцию, обменная кислотность не проявляется.
Водородные ионы поглощающего комплекса, вытесняемые гидролитически щелочными солями (образованными сильными основаниями и слабыми кислотами), обусловливают гидролитическую кислотность.
К гидролитически щелочным солям относятся, например, уксуснокислый натрий Ch4COONa и уксуснокислый кальций Са(СН3С00)2. Эти соли в водных растворах гидролитически расщепляются на слабую кислоту и сильное основание, которое придает раствору щелочную реакцию, при этом происходит более полное вытеснение поглощенного водорода.
Для определения гидролитической кислотности в лаборатории чаще всего используют уксуснокислый натрий.
Уксуснокислый натрий в воде гидролитически распадается на щелочь и слабую уксусную кислоту:
Ch4COONa + НОН = СН3СООН + NaOH.
При взаимодействии уксуснокислого натрия с подзолистой почвой натрий щелочи вступает в поглощающий комплекс и вытесняет поглощенный водород:
[почва] и + NaOH + СН3СООН = [почва] Na + Н,0 + СН3СООН.
Гидролитически щелочные соли вытесняют из поглощающего комплекса водорода больше, чем нейтральные соли. Поэтому наиболее полное представление о количестве поглощенных водородных ионов в почве можно получить на основании определения гидролитической кислотности.
Величину обменной и гидролитической кислотности почвы определяют титрованием солевой вытяжки щелочью (NaOH) и выражают в миллиграмм-эквивалентах на 100 г почвы. Обменную кислотность, кроме того, выражают в единицах рН путем определения концентрации водородных ионов в солевой вытяжке потенциомст-рически или колориметрически.
Почвенная кислотность неблагоприятна для развития растений и микроорганизмов и ведет к понижению плодородия почв. Устранение кислотности почв достигается известкованием, при котором происходит замещение поглощенного водорода на кальций:
Н г
[почва] и + Са (НС03)25: [почва] J? + 2НгО + 2С02. Н
Щелочность почвы. Почвы, в поглощающем комплексе которых находится натрий, имеют щелочную реакцию. Она обусловливается главным образом содой, образующейся в результате обмена поглощенного почвой натрия на водород углекислоты:
[почва] + Н2С03 ?> [почва] fj + Na2C03.
Широко распространен в природе и биологический процесс образования соды. Сульфатредуцирующие бактерии восстанавливают в анаэробных условиях в присутствии органического вещества сернокислые соли натрия до Na2S, который затем превращается в соду:
Na2S04 + 2С = 2С02 +Na2S;
Na2S + С02 + НгО = Na2C03 + HaS.
Высокая щелочность резко ухудшает физические и водные свойства почвы, усиливает пептизацию коллоидов, угнетает развитие растений, нарушая ход физиологических процессов.
Для устранения щелочности проводят гипсование:
[почва] [jj + CaS04it [почва] Са + Na, S04.
Сернокислый натрий легко вымывается из почвы, так как хорошо растворим в воде.
Почвенно-поглощающий комплекс (ППК)
Поглотительной способностью обладают коллоиды (частицы 0,2 – 0,001 мкм), предколлоидная фракция 1– 0,2 мкм. Характерной особенностью их является большая удельная поверхность. Это и определяет их высокую химическую активность.
Коллоиды подразделяются на
— минеральные
— органические
Часть минеральных коллоидов находится в кристаллическом состоянии. Это в основном минералы.
Другая часть минеральных коллоидов представляет собой аморфные вещества: к ним относят аллофаны, свежеосажденные гидраты полуторных оксидов ( Fe(OH)3, Al (OH)3, Mn (OH)3 ), гидраты кремнезема и их комплексные осадки (коагели).
Органическая часть почвенных коллоидов – аморфные гумусовые вещества, органо-минеральные комплексы, клетки мелких бактерий.
В большинстве почв преобладают минеральные коллоиды. Они составляют 85-90% их общей массы коллоидов. При физико-химической адсорбции поглощаются отдельные ионы (катионы и анионы). Адсорбция связана с наличием на поверхности коллоидных частиц положительного или отрицательного зарядов. Появление заряда на поверхности коллоидов, имеющих кристаллическое строение связано с некомпенсированностью зарядов ионов кристаллической решетки, расположенных на поверхности раздела твердая частица — раствор. Появление заряда в кристаллических и аморфных коллоидах, может быть обязано диссоциации ионов поверхностного слоя в окружающую среду.
3.3.2. Кислотность и основность в воде
Если воду подвергнуть многократной перегонке в кварцевой или платиновой посуде, то оказывается, что, несмотря на такую тщательную очистку, вода все же сохраняет небольшую, но вполне определенную способность проводить электрический ток. Это обусловлено самоионизацией воды:
 (3.8)
(3.8)
Константа равновесия (3.8) при строгой термодинамической записи имеет вид
 , (3.9)
, (3.9)
где  — относительная активность частицы Х в
равновесной смеси;
— относительная активность частицы Х в
равновесной смеси;  —
абсолютная активность Х в равновесной
смеси и
—
абсолютная активность Х в равновесной
смеси и — абсолютная активность Х в термодинамическом
состоянии системы, принятом за стандартное.
— абсолютная активность Х в термодинамическом
состоянии системы, принятом за стандартное.
Поскольку степень протекания
реакции (3.8) очень мала, относительная
активность самой воды при равновесии
будет очень мало отличаться от единицы
( ),
если в качестве стандартного состояния
взять гипотетическую чистую неионизированную
воду. Кроме того, поскольку равновесие
реакции (3.8) очень сильно сдвинуто влево,
коэффициенты активность ионов Н3О+ и ОН— в чистой воде будут также близки к
единице. Поэтому относительные активность
Н3О+ и ОН— фактически равны их молярным концентрациям
(если используется молярная шкала и в
качестве стандартного состояния выбрана
активность 1 моль/л). Отсюда
),
если в качестве стандартного состояния
взять гипотетическую чистую неионизированную
воду. Кроме того, поскольку равновесие
реакции (3.8) очень сильно сдвинуто влево,
коэффициенты активность ионов Н3О+ и ОН— в чистой воде будут также близки к
единице. Поэтому относительные активность
Н3О+ и ОН— фактически равны их молярным концентрациям
(если используется молярная шкала и в
качестве стандартного состояния выбрана
активность 1 моль/л). Отсюда
 ~Кавто=[H3O+][OH—],
~Кавто=[H3O+][OH—],
где [H3O+] и [OH—] — молярные концентрации; Кавто — константа автопротолиза воды, равная 1,00.10-14 моль2/л2 при 25оС.
Уравнение (3.8) показывает, что в чистой воде [H3O+] = [OH—], поэтому
[H3O+]
= [OH—]
=  = 10-7 при
25оС.
= 10-7 при
25оС.
Для удобства концентрацию гидроксониевых ионов целесообразно приводить в виде отрицательного логарифма, который обозначается символом рН:
рН = -lg[H3O+]
Для чистой воды рН = 7,00, в кислых растворах [H3O+]>[OH—] и рН<7, а в щелочных [H3O+]<[OH—] и рН>7.
Если [H3O+] или [OH—] нельзя считать очень малыми, то молярные концентрации уже не равны активностям, и нужно учитывать активность конкретного вида ионов.
Ниже для сравнения приведены константы автопротолиза некоторых других жидкостей (рКавто = -lg[Кавто] ) при 25оС.
Система | рКавто |
Серная кислота 2H2SO4 = H3SO4+ + HSO4— | 2,9 |
Муравьиная кислота 2HCOOH = HCOOH2+ + HCOO— | 6,2 |
Фтороводород 2HF = H2F+ + F— | 10,7 (0oC) |
Метанол 2CH3OH = CH3OH2+ + CH3O— | 16,7 |
Фторсульфоновая кислота 2FSO3H = FSO3H2+ + FSO3— | 17,4 |
Ацетонитрил 2CH3CN = CH3CNH+ + CH2=C=N— | 19,5 |
Аммиак 2NH3 = NH4+ + NH2— | 27,7 (-50oC) |
Диссоциацию кислоты АН в воде можно представить уравнением:
AH + H2O = A— + H3O+ (3.10)
В полном виде термодинамическая константа равновесия этой реакции выражается соотношением (ср. (3.9):
 ,
(3.11)
,
(3.11)
где  и
и — абсолютные активности частиц Х (Х=А—,
Н3О+,
АН, Н2О)
в равновесной смеси и в стандартном
состоянии соответственно.
— абсолютные активности частиц Х (Х=А—,
Н3О+,
АН, Н2О)
в равновесной смеси и в стандартном
состоянии соответственно.
Относительная активность
воды при состоянии равновесия ( )
не сильно изменяется при переходе от
одной кислоты у другой (для разбавленных
растворов) и при бесконечном разбавлении
приближается к единице. Поэтому можно
ввести так называемую термодинамическую
константу кислотности
)
не сильно изменяется при переходе от
одной кислоты у другой (для разбавленных
растворов) и при бесконечном разбавлении
приближается к единице. Поэтому можно
ввести так называемую термодинамическую
константу кислотности (АН):
(АН):
 (3.12)
(3.12)
Обычно используют шкалу
молярности и за стандартное состояние
принимают водный раствор, в котором
активность Х равна 1 моль/л при 25оС.
Таким образом, в уравнении (3.12) можно
сократить все размерности и член, в
который входит  .
Далее, если рассматривать процессы
только в разбавленных растворах, то
можно принять, что отношение коэффициентов
активности одинаково для всех кислот
и близко к единице. Тогда в качестве
относительной меры силы кислоты в
разбавленном водном растворе при данной
температуре можно использоватьконстанту
кислотности Ка(АН),
которая определяется выражением
.
Далее, если рассматривать процессы
только в разбавленных растворах, то
можно принять, что отношение коэффициентов
активности одинаково для всех кислот
и близко к единице. Тогда в качестве
относительной меры силы кислоты в
разбавленном водном растворе при данной
температуре можно использоватьконстанту
кислотности Ка(АН),
которая определяется выражением
Ка(АН)
=  , (3.13)
, (3.13)
где в квадратных скобках дана
молярная концентрация частиц в состоянии равновесия при
фиксированной температуре (обычно
25оС).
Конечно, Ка(АН)
будет зависеть от концентрации АН,
поскольку коэффициент активности связан
с концентрацией, но, если нужно,
термодинамический параметр  (АН)
можно получить экстраполяцией к
бесконечному разбавлению.
(АН)
можно получить экстраполяцией к
бесконечному разбавлению.
Чем больше Ка(АН), тем выше степень диссоциации (уравнение (3.10)) и тем сильнее кислота. Для характеристики кислотности удобно использовать отрицательный логарифм константы кислотности рКа(АН) или просто :рКа
рКа(АН) = -lgКа(АН). (3.14)
Очевидно, чем больше рКа, тем слабее кислота.
Из уравнений (3.12), (3.13) и (3.14) получаем
рКа(АН)
= рН — lg , (3.15)
, (3.15)
откуда следует что величина рКа равна тому значению рН раствора, при котором кислота ионизирована наполовину.
Для воды из уравнения (3.13) получаем
Ка= .
.
Таким образом, рКа(Н2О) = 15,7 при 25оС. Эта величина характеризует кислотность молекул воды в водном растворе. Для гидроксониевого иона рКа(Н3О+) = рКавто— pKa= 14-15,7 = -1,7.
Определение констант кислотности АН в воде с помощью прямого измерения концентраций А— и АН возможно лишь в том случае, если кислотная диссоциация происходит в заметной, но не полной степени. Если кислота так слаба, что почти не диссоциирует, или так сильна, что диссоциирует фактически нацело, то в первом случае нельзя точно измерить [A—], а во втором [AH]. Для таких кислот используются косвенные методы определения кислотности (разд. 3.3.4). В табл. 3.2 приведены величины рКа для некоторых кислот. Для кислот с рКа<0 или рКа>20 данные таблицы не очень точны. Однако некоторые значения определены с высокой степенью точности и экстраполированы к бесконечному разбавлению и поэтому представляют собой термодинамические величины рКао(НА) (уравнение (3.12)).
Соответствующее выражение для константы ионизации основания в воде можно получить аналогичным образом из уравнения

Это уравнение совершенно ясно показывает, что происходит, когда сильное основание растворяют в воде. Константа основности Кв определяется выражением
Кв= .
.
Однако в настоящее время константы основности практически вышли из употребления. Они просто излишни, так как информация о силе основания В включена в константу кислотности сопряженной кислоты ВН+:

Ка(ВН+)= .
.
Другими словами, один и тот же параметр — константа кислотности кислоты АН или ВН+ — является мерой не только силы АН или ВН+ как доноров протона, но и сила А— или В как акцепторов протона.
Сильной кислоте АН или ВН+ соответствует слабое сопряженное основание А— или В; в этом случае величина рКа мала или даже отрицательна. Сильному основанию А— или В соответствует слабая кислота АН или ВН+, в этом случае рКа имеет большое положительное значение. Так же как рН используется в качестве меры не только концентрации Н+, но и концентрации ОН—, так и рКа описывают не только силу А— или В как оснований, но и силу АН и ВН+ как кислот. В табл. 3.3 приведены значения рКа для некоторых оснований. Как и для кислот, непосредственно измерить силу оснований можно лишь в узком интервале рКа(ВН+). За пределами этого интервала основность определяется косвенными методами, поэтому значения рКа(ВН+) вне интервала примерно от -2 до 17 не являются точными.
Таблица 3.2
Основания функция кислотности — Справочник химика 21
Гордон изучал циклизацию латекса натурального каучука под действием 70—80%-ной серной кислоты в интервале температур 25—90°. Было показано, что скорости пропорциональны первой степени концентрации каучука и логарифму концентрации кислоты. Зависимость скорости от концентрации кислоты представляет собой зависимость, предсказываемую на основании функции кислотности Гаммета, которую можно применить к скорости образования сопряженного иона карбония (ВН+) из каучука (В). Таким образом [c.315]Хаммет предположил, что простая концентрация дает возможность экспериментально измерять степень протонизации в разбавленных растворах, соответствующий член в уравнении (XVI.5.4) можно использовать для измерения этого свойства в концентрированных растворах. Функцией кислотности растворов кислот по отношению к нейтральному основанию он назвал величину [c.494]
Гаммет предложил Г55] выражать кислотность с помощью так называемой нулевой функции кислотности. Яо, которая является мерой стремления протона кислоты перейти к незаряженной молекуле основания. [c.69]
При сравнении протонодонорной активности растворов заданной концентрации кислоты в различных растворителях оказывается, что функция кислотности Гаммета тем выше, чем ниже основность растворителя. Например, раствор НС1 в бензоле имеет значительно большую кислотность, чем в воде. Определяется это тем, что С1″ значительно более слабое основание, чем вода. Таким же образом растворы серной кислоты в-уксусной обладают значительно большей протонодонорной активностью (характеризуемой Но), чем растворы такой же концентрации в воде. В первом случае при внесении в раствор основания В устанавливается равновесие [c.160]
Основность углеводородов. Степень протонизации вещества в растворе кислоты с данной функцией кислотности Гаммета, как ясно из вышеизложенного, определяется основностью вещества, Для различных углеводородов, являющихся в общем очень слабыми основаниями, основность изменяется в очень широких пределах. Ниже приведены данные об основности некоторых аромати- [c.160]
Во-первых, соотношение коэффициентов активностей /в//вн зависит от природы основания В, и действительная функция кислотности для данного основания симбатна, но не равна и не строго пропорциональна функции кислотности Гаммета, построенной для оснований определенной химической природы (нитроанилинов). Во-вторых, на реакции с участием ионов очень Сильное влияние оказывают свойства среды (растворителя), в которой они протекают. В органической химии известны реакции, константы скорости которых в различных растворителях отличаются на 6—9 порядков. [c.162]
На основании исследования кислотно-основных равновесий в ледяной уксусной кислоте с помощью функции кислотности Гамметта, а также расчетов констант по данным Кольтгофа и Вильмана, Смит и Элиот в 1950 г. приняли, что при диссоциации образуются промежуточные продукты [c.299]
В дальнейшем были введены другие функции кислотности. В тех случаях, когда применяется в качестве индикатора незаряженная кислота и соответствующее ей основание имеет отрицательный заряд, функцию кислотности обозначают Я( ). [c.413]
Оказалось, что это далеко не так. Исследование э их функций кислотности в 0,002 н. растворе НС1 в смесях спирта с водой показало для функции Яо иную зависимость от содержания спирта, чем для функции Я( >. Это вполне естественно. В гл. УП были приведены многочисленные данные, согласно которым 70 для кислоты и ее аниона, безусловно, не равны между собой и их отношение не равно единице. Следовательно, Я( ) не передает истинной кислотности раствора. Появление заряда на молекуле основания в результате присоединения протона вызывает также резкое изменение энергии взаимодействия незаряженной молекулы индикатора и ее иона с растворителем. Все это говорит о том, что нельзя приравнивать изменение Яд к изменению кислотности. Задача может быть решена, если будут известны 7д для В и ВН+, только тогда Я о можно исправить и найти истинную [c.415]
Большинство функций кислотности применимы только к кислым растворам, однако известно несколько работ, посвященных сильно основным растворам. Так, функцию кислотности Я , применяемую к растворам сильных кислот, можно использовать также для растворов сильных оснований с зарядом, равным — 1 в этом случае она служит мерой способности растворителей отрывать протон от нейтральной кислоты ВН [76]. [c.334]
Однако точка максимального наклона кривой потенциометрического титрования достаточно часто не соответствует точке эквивалентности. Это происходит в тех случаях, когда определяемые ионы и ионы титранта имеют различные заряды, т е. стехиометрия реакции отличается от соотношения 1 Г Точка максимального наклона -образной кривой находится с той стороны от точки эквивалентности, где в избытке присутствует ион с меньшим зарядом. Ошибка титрования возрастает при увеличении произведения растворимости осадка в осадительном титровании, при уменьшении силы кислоты или основания в кислотно-основном титровании и при уменьшении прочности комплексов в комплексонометрическом титровании. Несовпадение точки эквивалентности и точки максимального наклона наблюдается также тогда, когда индикаторный электрод обратим лишь к одному из титруемых ионов или крутизна электродных функций к титруемому иону и иону-титранту различна. [c.248]
Для широкого круга слабых оснований в средах с высокими диэлектрическими проницаемостями приведенные равенства соблюдаются, и как уже показано выше, функцию кислотности можно выразить так [c.160]
Следуя Гаммету [177, гл. 9], примем для общего обозначения функции кислотности, безотносительно того, к какой сильной кислоте или к какому классу соединений она относится, символ Нх- Если используемая функция кислотности действительно описывает ионизацию данного вещества, то, согласно уравнениям (6.5) и (6.17), замена pH в формуле (6.9) на Нх позволяет непосредственно получать термодинамические константы Это дает возможность распространить описанную выше стандартную методику определения р а и на более слабые кислоты и основания. Так как функции Нх установлены с меньшей точностью, чем функция pH, при использовании функций кислотности допустимый размах в серии получаемых р/Сд может достигать 0,1 ед. [169, с. 76]. [c.123]
При работе со слабыми кислотами или основаниями (2 + в формулах (6.20) — (6.28) следует заменить на кх, для чего необходимо знать функцию кислотности, описывающую ионизацию данного вещества. К сожалению, при неизвестных Она и Од. исследователь не может проверить выбор функции кислотности, так как нельзя вычислить индикаторное отношение и оценить наклон прямолинейной зависимости 1 / = = /(Я ). Поэтому в случае неизвестных м. п. п. кислотно-основных форм следует попробовать находить их с использованием нескольких различных функций кислотности и окончательно остановиться на той из них, которая обеспечивает лучшее совпадение экспериментальной и вычисленной кривой В = 1(Нх). При использовании метода избыточной кислотности подбор требуемой Нх заменяется введением в задачу четвертого подбираемого параметра (л. ). [c.127]
Пример 8.7. при спектрофотометрическом определении основности некоторого амина по 7 точкам построена прямолинейная зависимость логарифма индикаторного отношения от функции кислотности Гаммета. Получено уравнение / = а + ЬНо с парам
кислотность основания — это… Что такое кислотность основания?
- кислотность основания
кислотность основания
—
[А.С.Гольдберг. Англо-русский энергетический словарь. 2006 г.]Тематики
- энергетика в целом
Справочник технического переводчика. – Интент. 2009-2013.
- кислотность органического удобрения
- кислотность пивного сусла [пива]
Смотреть что такое «кислотность основания» в других словарях:
Кислотность — Водородный показатель, pH (произносится «пэ аш»), это мера активности (в случае разбавленных растворов отражает концентрацию) ионов водорода в растворе, количественно выражающая его кислотность, вычисляется как отрицательный (взятый с обратным… … Википедия
Кислотность реакционной среды — Водородный показатель, pH (произносится «пэ аш»), это мера активности (в случае разбавленных растворов отражает концентрацию) ионов водорода в растворе, количественно выражающая его кислотность, вычисляется как отрицательный (взятый с обратным… … Википедия
Кислотность среды — Водородный показатель, pH (произносится «пэ аш»), это мера активности (в случае разбавленных растворов отражает концентрацию) ионов водорода в растворе, количественно выражающая его кислотность, вычисляется как отрицательный (взятый с обратным… … Википедия
Кислотность почв гидролитическая — способность п. связывать основания из растворов гидролитически щелочных солей. Определяется путем титрования щелочью 1 н. раствора ацетата Na (pH = 8,3) после однократного взаимодействия с ним навески п. и умножения полученной величины на… … Толковый словарь по почвоведению
КИСЛОТЫ И ОСНОВАНИЯ — Термины кислоты и основания вполне сформировались в 17 в. Их содержание неоднократно пересматривалось и дополнялось. Этот процесс происходил и происходит в острых столкновениях представителей разных взглядов на природу К. и о. Развитие взглядов… … Химическая энциклопедия
ЭКВИВАЛЕНТНОЕ КОЛИЧЕСТВО ВЕЩЕСТВА — физ. величина, равная для элемента кол ву в ва, соединяющемуся с атомарным водородом или замещающему его в хим. соединениях. Единица Э, к. в. (в СИ) моль. Э. к. в. для к ты равно кол ву в ва, делённому на основность к ты (число ионов водорода),… … Большой энциклопедический политехнический словарь
МОЧА — (урина, urina), жидкость, отде ляемая почками и выделяемая из организ ма наружу через систему мочевыводящих путей. СМ. удаляются из организма почти все азотистые продукты обмена веществ (за исключением небольших количеств, поступающих в пот и в… … Большая медицинская энциклопедия
Спирты — Отличительная особенность спиртов гидроксильная группа при насыщенном атоме углерода на рисунке выделена красным (кислород) и серым цветом (водород). Спирты (от лат. … Википедия
ЖЕЛУДОК — ЖЕЛУДОК. (gaster, ventriculus), расширенный отдел кишечника, имеющий благодаря наличию специальных желез значение особо важного пищеварительного органа. Ясно диференцированные «желудки» многих беспозвоночных, особенно членистоногих и… … Большая медицинская энциклопедия
Теории кислот и оснований — совокупность фундаментальных физико химических представлений, описывающих природу и свойства кислот и оснований. Все они вводят определения кислот и оснований двух классов веществ, реагирующих между собой. Задача теории предсказание продуктов… … Википедия