Кислотно-основные свойства соединений
Тема30.МЕТОДМОЛЕКУЛЯРНЫХОРБИТАЛЕЙ
Основная идея метода молекулярных орбиталей (МО) заключа-
ется в том, что все электроны находятся на молекулярных орбиталях, единых для всей системы ядер и электронов данной молекулы. Для определения волновых функций молекулярных орбиталей используют приближение, называемое линейной комбинацией атомных орбиталей (ЛКАО): молекулярная волновая функция МО получается простым сложением или вычитанием атомных волновых функций АО. Основные положения метода МО ЛКАО (метода молекулярных орбиталей в предположении линейной комбинации атомных орбиталей) заключаются в следующем:
–в образовании МО могут участвовать АО, имеющие сходную симметрию и близкую энергию;
–количество МО в молекуле равно сумме АО атомов, входящих в
еесостав;
–АО должны в достаточной степени перекрываться при образовании МО. Поэтому орбитали внутренних электронных слоев атомов практически не участвуют в образовании связи и в методе МО обычно не рассматриваются.
По аналогии с атомными s-, p-, d-, f-орбиталями молекулярные
орбитали обозначают греческими буквами σ, π, δ, ϕ. Заполнение молекулярных орбиталей происходит так же, как и атомных, с соблюдением принципа Паули, принципа наименьшей энергии и правила Хунда; на каждой МО может находиться не более двух электронов.
В методе МО образование молекулярных орбиталей рассматривается как результат сложения и вычитания комбинируемых АО. Молекулярная орбиталь, возникающая в результате сложения АО, отвечает более низкому значению энергии, чем сумма энергий исходных АО. Такая МО имеет повышенную электронную плотность в пространстве между ядрами и способствует образованию химической связи, она на-
зывается связывающей.
Молекулярная орбиталь, возникающая в результате вычитания атомных орбиталей, отвечает более высокому значению энергии, чем сумма энергий исходных АО. Электронная плотность в этом случае сконцентрирована за ядрами атомов, а между ними равна нулю. Подобные МО энергетически менее устойчивы, они приводят к ослаблению химической связи и называются разрыхляющими (их часто обозначают звездочками на схемах).
Кислотно-основные свойства соединений
Тема30.МЕТОДМОЛЕКУЛЯРНЫХОРБИТАЛЕЙ
Основная идея метода молекулярных орбиталей (МО) заключа-
ется в том, что все электроны находятся на молекулярных орбиталях, единых для всей системы ядер и электронов данной молекулы. Для определения волновых функций молекулярных орбиталей используют приближение, называемое линейной комбинацией атомных орбиталей (ЛКАО): молекулярная волновая функция МО получается простым сложением или вычитанием атомных волновых функций АО. Основные положения метода МО ЛКАО (метода молекулярных орбиталей в предположении линейной комбинации атомных орбиталей) заключаются в следующем:
–в образовании МО могут участвовать АО, имеющие сходную симметрию и близкую энергию;
–количество МО в молекуле равно сумме АО атомов, входящих в
еесостав;
–АО должны в достаточной степени перекрываться при образовании МО. Поэтому орбитали внутренних электронных слоев атомов практически не участвуют в образовании связи и в методе МО обычно не рассматриваются.
По аналогии с атомными s-, p-, d-, f-орбиталями молекулярные
орбитали обозначают греческими буквами σ, π, δ, ϕ. Заполнение молекулярных орбиталей происходит так же, как и атомных, с соблюдением принципа Паули, принципа наименьшей энергии и правила Хунда; на каждой МО может находиться не более двух электронов.
В методе МО образование молекулярных орбиталей рассматривается как результат сложения и вычитания комбинируемых АО. Молекулярная орбиталь, возникающая в результате сложения АО, отвечает более низкому значению энергии, чем сумма энергий исходных АО. Такая МО имеет повышенную электронную плотность в пространстве между ядрами и способствует образованию химической связи, она на-
зывается связывающей.
Молекулярная орбиталь, возникающая в результате вычитания атомных орбиталей, отвечает более высокому значению энергии, чем сумма энергий исходных АО. Электронная плотность в этом случае сконцентрирована за ядрами атомов, а между ними равна нулю. Подобные МО энергетически менее устойчивы, они приводят к ослаблению химической связи и называются разрыхляющими (их часто обозначают звездочками на схемах).
14. Кислотно-основное равновесие. Типы протолитических реакций.
Согласно ионной теории кислоты – соединения, которые при электролитической диссоциации в водном растворе образуют ионы водорода Н+:
НАn 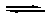 Н+ + Аn−
Н+ + Аn−
Основания – соединения, которые при электролитической диссоциации в водном растворе образуют ионы гидроксила ОН−:
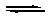 Kt+ + ОН−
Kt+ + ОН−Рассматривая кислотно-основные равновесия в водных растворах в дальнейшем, будем считать их приближающимися к идеальным растворам, т.е. активностью ионов пренебрегаем (а → С).
Тогда в соответствии с законом действующих масс:
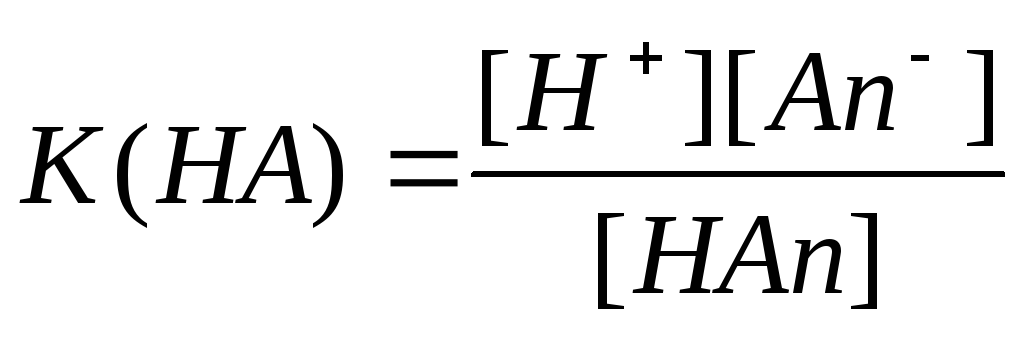

Типы протолитической реакции:
Реакция нейтрализации. Кислота и щелочь в растворе обмениваются ионами и нейтрализуют друг друга.
NaOH + HCl → NaCl + H2O
Реакция гидролиза.
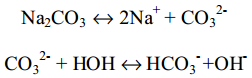
15. Кислотные и основные свойства растворителей. Влияние природы растворителя на силу кислот и оснований. Константа кислотности и основности. Нивелирующее и дифференцирующее действие растворителей.
С точки зрения кислотно-основных свойств растворители можно разделить на три группы:
1. Апротонные растворители, не обладающие ни кислотными, ни основными свойствами — например, углеводороды и их галогенопроизводные: бензол, гексан, хлороформ.
2. Протофильные растворители, обладающие только основными свойствами: кетоны (ацетон), простые эфиры (диоксан, диэтиловый эфир), третичные амины (пиридин).
3. Амфипротные растворители, обладающие как кислотными, так и основными свойствами: вода, спирты, карбоновые кислоты, первичные и вторичные амины.
Диссоциация слабых кислот. CH3COOH ↔ CH3COO— + H+
Ka — константа равновесия, называемая константой кислотности
Диссоциация слабых оснований. NH4OH ↔ NH4+ + OH—
Kb-константа равновесия, называемая константой основности
Чем больше Ка и Kb, тем сильнее диссоциируют кислоты и основания в водных растворах.
Важная роль растворителя в кислотно-основных реакциях ярко проявляется в нивелирующем и дифференцирующем эффектах.
В области высоких (Ка > 1) и очень низких (Ка < 10-14) значений констант кислотности величина рН практически не зависит от рКа (константа ионизации). Аналогичные явления наблюдаются и для водных растворов оснований, а также для растворов кислот и оснований в любом другом амфипротном растворителе. Подобная независимость кислотно-основных свойств от констант кислотности (основности) называется нивелирующий эффектом растворителя.
При переходе от более к менее основному растворителю происходит обратное явление: сильные кислоты могут стать слабыми. Например, в воде НС1 и НСlO
4 — сильные кислоты. В ледяной уксусной кислоте они становятся слабыми и, следовательно, различающимися по своим кислотно-основным свойствам. Этот эффект называется дифференцирующим эффектом растворителя. Дифференцирующий эффект проявляется и для оснований — при переходе от более к менее кислотному растворителю (например, от воды — к пиридину).Как видно, в кислотно-основных равновесиях растворитель играет не меньшую роль, чем природа кислот и оснований. С точки зрения равновесия из многочисленных свойств растворителей имеет значение донорно-акцепторное сродство к протону и диэлектрическая проницаемость.
Влияние структурных факторов на относительную силу кислот и оснований
Сила кислоты или основания определяется положением равновесия кислотно-основного взаимодействия и зависит от разности свободных энергий исходных и конечных соединений. Поэтому факторы, которые стабилизируют сопряженное основание в большей степени, чем кислоту, увеличивают кислотность и уменьшают основность. Факторы, стабилизирующие преимущественно кислоту по сравнению с основанием, действуют в противоположном направлении. Поскольку сопряженные основания, как правило, несут отрицательный заряд, то увеличению кислотности способствуют факторы, стабилизирующие анион.
Влияние строения на силу кислот и оснований.
Сила кислоты зависит от природы атома при кислотном центре и от его структурного окружения.
Для оценки относительной силы кислот важны такие характеристики атома при кислотном центре как его электроотрицательность и поляризуемость.
При прочих равных условиях для элементов одного периода с ростом электроотрицательности атома кислотность соединений увеличивается, так как высокая электроотрицательность атома при кислотном центре стабилизирует образующийся при отщеплении протона анион. Так, кислотность уменьшается в ряду:
OH-кислоты> NH-кислоты> CH-кислоты
| CH3O-H | CH3NH-H | CH3CH2-H |
pKa | 16 | 30 | 40 |
Электроотрицательность атома зависит не только от его природы, но и от типа гибридизации и возрастает по мере увеличения s-характера гибридных орбиталей. Параллельно возрастает кислотность соединений:
| СH3CH | CH2=CH-H |
|
pKa | 40 | 36 | 25 |
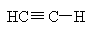
Для элементов одной подгруппы с возрастанием заряда ядра кислотность соединений увеличивается:
OH-кислоты < SH-кислоты
| CH3O-H | CH3S-H |
pKa | 16,0 | 10,5 |
Увеличение кислотности соединений, несмотря на снижение электроотрицательности атомов в подгруппе, связано с увеличением их поляризуемости по мере возрастания радиуса атома. Большая поляризуемость атома способствует лучшей делокализации отрицательного заряда и повышению стабильности сопряженного основания.
При одинаковой природе атома при кислотном центре сила кислоты определяется его структурным окружением. Увеличению силы кислоты способствует делокализация отрицательного заряда в сопряженном основании (анионе) и его рассредоточение на большем количестве атомов.
Так, карбоновые кислоты – одни из самых сильных органических кислот. Их сила обусловлена стабилизацией карбоксилат-аниона за счет делокализации отрицательного заряда в сопряженной системе. В результате отрицательный заряд в карбоксилат-анионе рассредоточен между двумя атомами кислорода, а обе связи С-O абсолютно равноценны:

Фенолы являются более сильными кислотами, чем спирты, за счет резонансной стабилизации фенолят-аниона, отрицательный заряд которого делокализован по ароматическому кольцу:
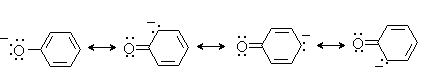
В результате по силе органические OH-кислоты могут быть расположены в следующий ряд:
| ROH | < | H2O | < | ArOH | < | RCOOH |
pKa | 16-17 |
| 15,7 |
| 8-11 |
| 4-5 |
Введение заместителя в связанный с кислотным центром углеводородный радикал влияет на силу кислоты. Электроноакцепторные(ЭА) заместители увеличивают, а электронодонорные(ЭД) — уменьшают кислотность. Влияние электроноакцепторных заместителей связано с их способностью делокализовать отрицательный заряд и, тем самым стабилизировать сопряженное основание (анион). Влияние электронодонорных заместителей приводит к дестабилизации аниона.
Электроноакцепторные заместители увеличивают силу алифатических и ароматических карбоновых кислот, электронодонорные заместители действуют в противоположном направлении:
| Cl-CH2-COOH | H-COOH | CH3-COOH |
pKa | 2,8 | 3,7 | 4,7 |
|
|
| |
| +M > -I |
| -M и –I |
pKa | 4,47 | 4,20 | 3,43 |
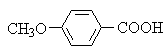
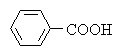

Аналогичное влияние оказывают заместители на кислотность спиртов и фенолов.
При одинаковом структурном окружении для элементов одного периода с ростом электроотрицательности атома при основном центре основность соединений уменьшается:
аммониевые основания > оксониевые основания
| ROH | RNH2 |
| ~2 | ~10 |
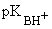
Снижение основности связано с тем, что более электроотрицательный атом прочнее удерживает неподеленную пару электронов, которую он должен отдать на образование связи с протоном.
Увеличение s-характера гибридных орбиталей приводит к снижению основности:

Для элементов одной подгруппы с возрастанием заряда ядра основность уменьшается:
оксониевые основания > сульфониевые основания
Введение электронодонорных заместителей увеличивает, а введение электроакцепторных — понижает основность. Так, электронодонорные заместители увеличивают основность алифатических и ароматических аминов, увеличивая склонность электронной пары азота к атаке протона. Электроноакцепторные заместители напротив снижают электронную плотность неподеленной пары электронов азота и делают ее менее восприимчивой для атаки протоном:
Если свободная пара электронов азота находится в сопряжение с двойной связью или ароматическим кольцом, основность снижается. Так, в анилине свободная пара электронов азота сопряжена с ароматическим кольцом.
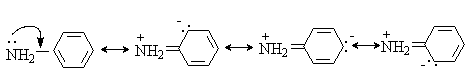
Протонирование анилина приводит к нарушению сопряжения и энергетически менее выгодно, чем протонирование алифатических аминов.
Амиды карбоновых кислот являются очень слабыми основаниями из-за сопряжения пары электронов азота с карбонильной группой. В результате атом азота приобретает частичный положительный, а атом кислорода – частичный отрицательный заряд, и протонирование амидов происходит, как правило, по атому кислорода.
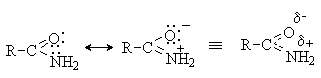
Основность азотсодержащих гетероциклических соединений также определяется доступностью пары электронов азота для атаки протона. Высокой основностью обладают насыщенные азотсодержащие гетероциклы, в которых атом азота находится в состоянии sp3-гибридизации. Основность пиридиниевого атома азота (sp2-гибридизация) ниже. Наконец, пиррольный атом азота практических лишен основных свойств, так как его протонирование означает разрушение ароматической гетероциклической системы: