© ГБПОУ КК ПАТИС
ГБПОУ КК ПАТИС
Государственное бюджетное профессиональное образовательное учреждение Краснодарского края
Приморско-Ахтарский техникум индустрии и сервиса
Адрес: 353860 г. Приморско-Ахтарск, ул. Тамаровского, 85
тел: 8 (861-43) 2-35-94, 8 (861-43) 2-18-98
Адрес сайта: http://патис.рф
Социальные сети: VK и OK
Электронная почта: [email protected]
Режим работы:
ПН — СБ: с 8.00 до 16.00
Выходные дни: ВС
Учредители
Наименование:
Министерство образования, науки и молодежной политики Краснодарского края
Адрес: 350063 г. Краснодар, ул. Рашпилевская, 23
тел: 8 (861) 298-25-73
Адрес сайта: minobr. krasnodar.ru
krasnodar.ru
Электронная почта: [email protected]
Режим работы:
ПН.ВТ.СР.ЧТ. – с 09.00 до 18.00
ПТ. – с 09.00 до 17.00
Перерыв на обед: с 13.00 до 13.50
Выходные дни: СБ.ВС.
Наименование:
Департамент имущественных отношений Краснодарского края
Адрес: 350000 г. Краснодар, ул. Гимназическая, 36
Канцелярия: 8 (861) 268-24-08
Факс: 8 (861) 267-11-75
Специалист по работе с обращениями граждан — консультации, запись на прием — телефон 267-11-78
Телефон горячей линии по вопросам земельных отношений: 8 (861) 992-33-35
Адрес сайта: diok.krasnodar.ruЭлектронная почта: [email protected]
Режим работы:
ПН. ВТ.СР.ЧТ. – с 09.00 до 18.00
ВТ.СР.ЧТ. – с 09.00 до 18.00
ПТ. – с 09.00 до 17.00
Перерыв на обед ПН.ВТ.СР.ЧТ.: с 13.00 до 13.50
Перерыв на обед ПТ.: с 13.00 до 13.40
Выходные дни: СБ.ВС.
Теория химического строения органических соединений. Электронная природа химических связей. Предпосылки теории строения. Теория химического строения. Изомерия реферат по химии
БАШКИРСКИЙ ЭКОНОМИКО-ЮРИДИЧЕСКИЙ ТЕХНИКУМ Курсовая работа ПО ДИСЦИПЛИНЕ «ОРГАНИЧЕСКАЯ ХИМИЯ» НА ТЕМУ: «ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ. ЭЛЕКТРОННАЯ ПРИРОДА ХИМИЧЕСКИХ СВЯЗЕЙ, ПРЕДПОСЫЛКИ ТЕОРИИ СТРОЕНИЯ. ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ. ИЗОМЕРИЯ» Выполнил студент: Очного отделения Юридического факультета Группы О-05-19 Диргамов Р.Р. Проверила преподаватель: Исламгулова. И.М. с. Иванаево-2008 год СОДЕРЖАНИЕ 1. ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ. ЭЛЕКТРОННАЯ ПРИРОДА ХИМИЧЕСКИХ СВЯЗЕЙ, ПРЕДПОСЫЛКИ ТЕОРИИ СТРОЕНИЯ…………………….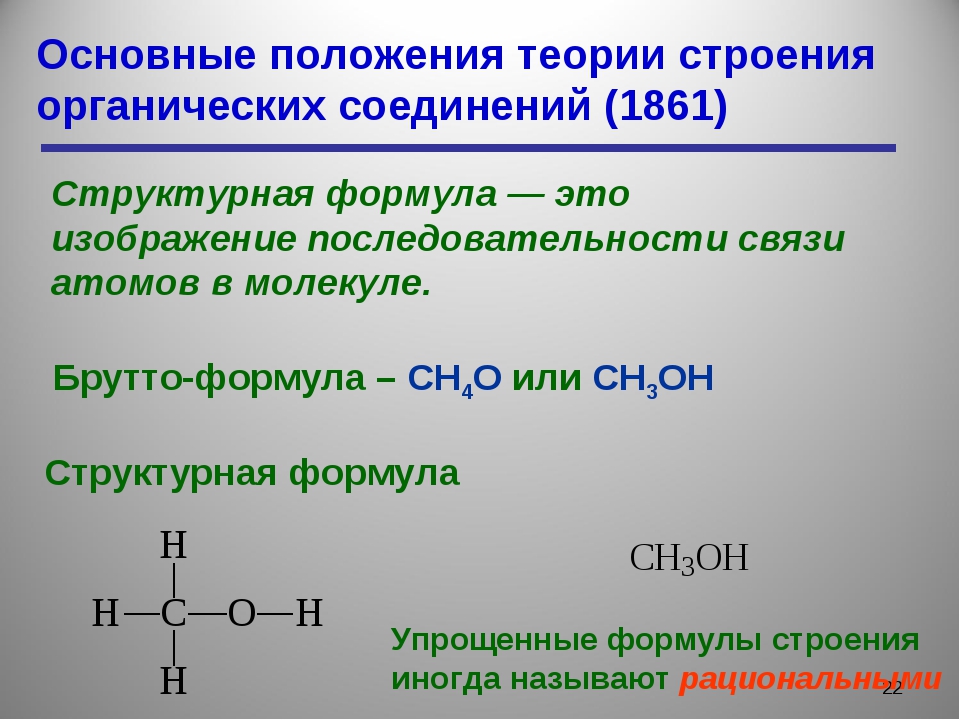 .………..…СТР. 7 2. ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ…………………………….…СТР. 9 3. ЗАКЛЮЧЕНИЕ……………………………………………………..…..СТР. 13 4. СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ……………..…..…СТР. 14 ученый А.М. Вутлеров в 1861г. впервые синтезом получает сахаристое вещество. Синтезы веществ, ранее вырабатывавшихся только живыми организмами, начали быстро следовать один за другим. Идеалистическое учение о «жизненной силе потерпело полное поражение. В настоящее время синтезированные многие органические вещества, не только имеющиеся в природе, но и не встречающиеся в ней, например: многочисленные пластмассы, различные виды каучуков, всевозможные красители, взрывчатые вещества, лекарственные препараты. Синтетически полученных веществ сейчас известно даже больше, чем найденных в природе, и число их быстро растет. Начинают осуществляться синтезы самых сложных органических веществ — белков. Смысл термина «органические вещества» давно стал шире его первоначального значения. Теперь это название охватывает не только вещества, входящие в состав организмов, но и синтетически получаемые, не имеющие отношения к организмам.
.………..…СТР. 7 2. ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ…………………………….…СТР. 9 3. ЗАКЛЮЧЕНИЕ……………………………………………………..…..СТР. 13 4. СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ……………..…..…СТР. 14 ученый А.М. Вутлеров в 1861г. впервые синтезом получает сахаристое вещество. Синтезы веществ, ранее вырабатывавшихся только живыми организмами, начали быстро следовать один за другим. Идеалистическое учение о «жизненной силе потерпело полное поражение. В настоящее время синтезированные многие органические вещества, не только имеющиеся в природе, но и не встречающиеся в ней, например: многочисленные пластмассы, различные виды каучуков, всевозможные красители, взрывчатые вещества, лекарственные препараты. Синтетически полученных веществ сейчас известно даже больше, чем найденных в природе, и число их быстро растет. Начинают осуществляться синтезы самых сложных органических веществ — белков. Смысл термина «органические вещества» давно стал шире его первоначального значения. Теперь это название охватывает не только вещества, входящие в состав организмов, но и синтетически получаемые, не имеющие отношения к организмам. Однако, как исторически сложившееся, это название оставлено для обозначения всей многочисленной группы веществ, содержащих углерод. Название науки «органическая химия», утратив первоначальный смысл, приобрело в связи с этим более широкое толкование. Можно сказать, что такое название получило и новое подтверждение, так как ведущей познавательной задачей современной органической химии является глубокое изучение процессов, происходящих в клетках организмов на молекулярном уровне, выяснение тех тонких механизмов, которые составляют материальную основу явлений жизни. Изучение химии органических веществ, таким образом, расширяет наши знания о природе. Раскрывая взаимосвязь веществ, прослеживая процесс усложнения их от наиболее простых — неорганических — до самых сложных, составляющих организмы, эта наука раскрывает нам картину развития природы, позволяет глубже понять процессы, происходящие в природе, и закономерности, лежащие в их основе. Достижения органической химии широко используются в современном производстве.
Однако, как исторически сложившееся, это название оставлено для обозначения всей многочисленной группы веществ, содержащих углерод. Название науки «органическая химия», утратив первоначальный смысл, приобрело в связи с этим более широкое толкование. Можно сказать, что такое название получило и новое подтверждение, так как ведущей познавательной задачей современной органической химии является глубокое изучение процессов, происходящих в клетках организмов на молекулярном уровне, выяснение тех тонких механизмов, которые составляют материальную основу явлений жизни. Изучение химии органических веществ, таким образом, расширяет наши знания о природе. Раскрывая взаимосвязь веществ, прослеживая процесс усложнения их от наиболее простых — неорганических — до самых сложных, составляющих организмы, эта наука раскрывает нам картину развития природы, позволяет глубже понять процессы, происходящие в природе, и закономерности, лежащие в их основе. Достижения органической химии широко используются в современном производстве. Осуществляя в широких масштабах процессы переработки природных веществ и разнообразные органические, промышленность органической химии создает многочисленные вещества и для других отраслей промышленные кости, с/х. культуры, быта. Все эти стороны органической химии раскроются перед вами в проессе дальнейшего изучения науки. Глава 1. ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ ОРГАНИЧЕСКИХ Мы отметили лишь несколько проблем, требовавших теоретического объяснения. Перед учеными того времени стояли и другие сложные вопросы. 2. ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ Основы новой теории сформулировован 1861г. профессор Казанского университета Александр Бутлеров. В химии к тому времени уже значительное распространение получили идеи атомистики. Понят атома и молекулы получили на международном съезде Химиков в 1860г. свое точное определение. Но ученые еще не придавали значения тому, как строятся молекулы из атомное, и считали, что позвать это строение химическими методами невозможно. Были и такие ученые, которые вообще не реального существования атомов и молекул.
Осуществляя в широких масштабах процессы переработки природных веществ и разнообразные органические, промышленность органической химии создает многочисленные вещества и для других отраслей промышленные кости, с/х. культуры, быта. Все эти стороны органической химии раскроются перед вами в проессе дальнейшего изучения науки. Глава 1. ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ ОРГАНИЧЕСКИХ Мы отметили лишь несколько проблем, требовавших теоретического объяснения. Перед учеными того времени стояли и другие сложные вопросы. 2. ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ Основы новой теории сформулировован 1861г. профессор Казанского университета Александр Бутлеров. В химии к тому времени уже значительное распространение получили идеи атомистики. Понят атома и молекулы получили на международном съезде Химиков в 1860г. свое точное определение. Но ученые еще не придавали значения тому, как строятся молекулы из атомное, и считали, что позвать это строение химическими методами невозможно. Были и такие ученые, которые вообще не реального существования атомов и молекул. А.М. Бутлеров не только считал атомы и молекулы реально существующими частицами веществ, но и пришел к выводу, что атомы в молекулах не находится в беспорядке, а соединены друг с другом в определенной последовательности которую можно установить химическими методами и отразить в формуле. Основную идею своей теории А.М. Бутлеров выразил в следующих словах: е Химическая натура сосной частицы определяется натурой элементарных составных частей, количеством их и химическим строением». В более привычной для нас формулировке это означает, что химические свойства молекулы определяются свойствами составляющих ее атомов, их числом и химическим строением молекулы, Химическое строение, по А.М. Бутлерову — это последовательность соединения атомов в молекуле, порядок их взаимосвязи и взаимного влияния друг Руга. Соединения атомов в молекулы, указывал А.М. Бутлеров происходит в соответствии с их атомностью (валентностью). На примерах из неорганической химии можно видеть, что атомы, соединяясь в Молекулу, оказывают влияние друг на друга.
А.М. Бутлеров не только считал атомы и молекулы реально существующими частицами веществ, но и пришел к выводу, что атомы в молекулах не находится в беспорядке, а соединены друг с другом в определенной последовательности которую можно установить химическими методами и отразить в формуле. Основную идею своей теории А.М. Бутлеров выразил в следующих словах: е Химическая натура сосной частицы определяется натурой элементарных составных частей, количеством их и химическим строением». В более привычной для нас формулировке это означает, что химические свойства молекулы определяются свойствами составляющих ее атомов, их числом и химическим строением молекулы, Химическое строение, по А.М. Бутлерову — это последовательность соединения атомов в молекуле, порядок их взаимосвязи и взаимного влияния друг Руга. Соединения атомов в молекулы, указывал А.М. Бутлеров происходит в соответствии с их атомностью (валентностью). На примерах из неорганической химии можно видеть, что атомы, соединяясь в Молекулу, оказывают влияние друг на друга. Так, водород и кислород, образовав воду, настолько изменились в результате взаимного влияния, что первый уже Не Роит, а второй не поддерживает горения; вода не Обещает свойствами ни водорода, ни кислорода. Основывается на приведенных выше высказываниях А. М. Бутлерова, сущность теории химического строения можно выразить в следующих положениях: • 1. Атомы в молекулах располагаются не беспорядочно, они соединены друг с другом в определенной последовательности согласно их валентности. • 2. Свойства веществ зависят не только от того, атомы каких элементов и в каком количестве входят в состав молекул, но и от последовательности соединения атомов в молекулах, от порядка их взаимного влияния друг на друга. Рассмотрим на примере известных нам углеводородов первое из этих положений. В какой последовательности соединены атомы в молекуле простейшего углеводорода метана? Мы уже знаем, что каждый атом водорода в нем соединен с атомом углерода. Легко понять, что иначе и быть не может. Если, например, предположить, что какие-нибудь два атома водорода связаны друг с другом непосредственно, то, исчерпав при этом свою валентность, они уже не смогут соединяться с другими атомами.
Так, водород и кислород, образовав воду, настолько изменились в результате взаимного влияния, что первый уже Не Роит, а второй не поддерживает горения; вода не Обещает свойствами ни водорода, ни кислорода. Основывается на приведенных выше высказываниях А. М. Бутлерова, сущность теории химического строения можно выразить в следующих положениях: • 1. Атомы в молекулах располагаются не беспорядочно, они соединены друг с другом в определенной последовательности согласно их валентности. • 2. Свойства веществ зависят не только от того, атомы каких элементов и в каком количестве входят в состав молекул, но и от последовательности соединения атомов в молекулах, от порядка их взаимного влияния друг на друга. Рассмотрим на примере известных нам углеводородов первое из этих положений. В какой последовательности соединены атомы в молекуле простейшего углеводорода метана? Мы уже знаем, что каждый атом водорода в нем соединен с атомом углерода. Легко понять, что иначе и быть не может. Если, например, предположить, что какие-нибудь два атома водорода связаны друг с другом непосредственно, то, исчерпав при этом свою валентность, они уже не смогут соединяться с другими атомами. Обозначая валентность элементов условно черточками, мы так изображаем порядок связи атомов в молекуле метана: В молекулах пропана С3Н3 и бутана С4Н10 атомы соединены в таком порядке: Зная строение углеводородов, мы теперь можем ответить на некоторые из тех вопросов, которые волновали в свое время ученых.
Обозначая валентность элементов условно черточками, мы так изображаем порядок связи атомов в молекуле метана: В молекулах пропана С3Н3 и бутана С4Н10 атомы соединены в таком порядке: Зная строение углеводородов, мы теперь можем ответить на некоторые из тех вопросов, которые волновали в свое время ученых.
Классическая теория химического строения
В 1857-1858 гг. по вопросам химического строения молекул высказывались многие ученые: в России — А.М.Бутлеров (1828-1886), шотландский химик А.Купер (1831-1892), немецкий химик А.Кекуле (1829-1896) и др. Кекуле писал: «Определенные идеи в определенное время буквально «носятся в воздухе», и если один человек не высказал возникшую у него идею, она будет очень скоро высказана другим».
Изучая металлорганические
соединения английский химик
Э. Франкланд (1825-1899) пришел к
выводу, что металлы в этих
соединениях проявляют определенную
«емкость насыщения». Термин
«валентность» появился лишь в 1868 г.
Как синонимы понятию «валентность»
широко использовались термины
«атомность», «единица сродства»,
«сумма химических единиц элементов»,
«степень заместимости» и т.д. К 1858 г.
было установлено, что валентность
водорода во всех соединениях всегда
равна единице, валентность хлора в
органических соединениях также
равна единице, валентность кислорода
и серы равна двум, валентность
углерода равна четырем. Так А.Кекуле
в 1858 г. писал: «При рассмотрении
простейших соединений углерода
(рудничный газ, хлористый метил,
хлористый углерод, хлороформ,
угольная кислота, фосгеновый газ,
сероуглерод, синильная кислота и т.д.)
бросается в глаза, что количество
углерода, которое химики считают
наименьшим из возможных и признают
атомом, всегда связывает четыре атома
одноатомного или два атома
двухатомного элемента, что вообще
сумма химических единиц элемента,
связанных с атомом углерода, равна 4.
Франкланд (1825-1899) пришел к
выводу, что металлы в этих
соединениях проявляют определенную
«емкость насыщения». Термин
«валентность» появился лишь в 1868 г.
Как синонимы понятию «валентность»
широко использовались термины
«атомность», «единица сродства»,
«сумма химических единиц элементов»,
«степень заместимости» и т.д. К 1858 г.
было установлено, что валентность
водорода во всех соединениях всегда
равна единице, валентность хлора в
органических соединениях также
равна единице, валентность кислорода
и серы равна двум, валентность
углерода равна четырем. Так А.Кекуле
в 1858 г. писал: «При рассмотрении
простейших соединений углерода
(рудничный газ, хлористый метил,
хлористый углерод, хлороформ,
угольная кислота, фосгеновый газ,
сероуглерод, синильная кислота и т.д.)
бросается в глаза, что количество
углерода, которое химики считают
наименьшим из возможных и признают
атомом, всегда связывает четыре атома
одноатомного или два атома
двухатомного элемента, что вообще
сумма химических единиц элемента,
связанных с атомом углерода, равна 4.
В 1861 г. на собрании Общества
немецких естествоиспытателей и
врачей в Шпайере выступил с докладом
Александр Михайлович Бутлеров. Он
изложил свою теорию химического
строения: «Все сознают, что
теоретическая сторона химии не
соответствует ее фактическому
развитию. …В настоящее время лишь
атомность составляет определенное и
неизменное свойство элементов и
может служить основанием общей
теории. …Химическая натура сложной
частицы определяется натурой
элементарных составных частей,
количеством их и химическим
строением. …Если теперь определить
химическое строение веществ и
выразить его рациональными
формулами, то для каждого тела
возможна будет лишь одна
рациональная формула».
А.М.Бутлеров ввел понятие «взаимное
влияние атомов в молекуле
непосредственно не связанных
химическими связями». Эту идею
плодотворно развил его ученик
В.В.Марковников. Он синтезировал
изомасляную кислоту (существование
которой было предсказано теорией
химического строения), установил
отличие ее свойств от свойств
нормальной масляной кислоты.
В 1865 г. А.Кекуле предложил свою
структурную формулу для молекулы
бензола , а в 1866 г.
Эрленмейер, имея в виду родство в
химических свойствах бензола и
нафталина, предложил для молекулы
нафталина формулу, состоящую из двух
бензольных колец с двумя общими
углеродными атомами. Но не все
ученые-химики принимали и развивали
теорию химического строения, были у
нее и яростные противники. Так,
руководитель обширной химической
лаборатории в Лейпцигском
университете (Германия), талантливый
химик-экспериментатор, главный
редактор «Журнала практической
химии» Г.Кольбе (1818-1884) в 1870 г.
писал, что теория химического
строения это просто «игра для
химических детей», у которых
«извращенные представления» о химии.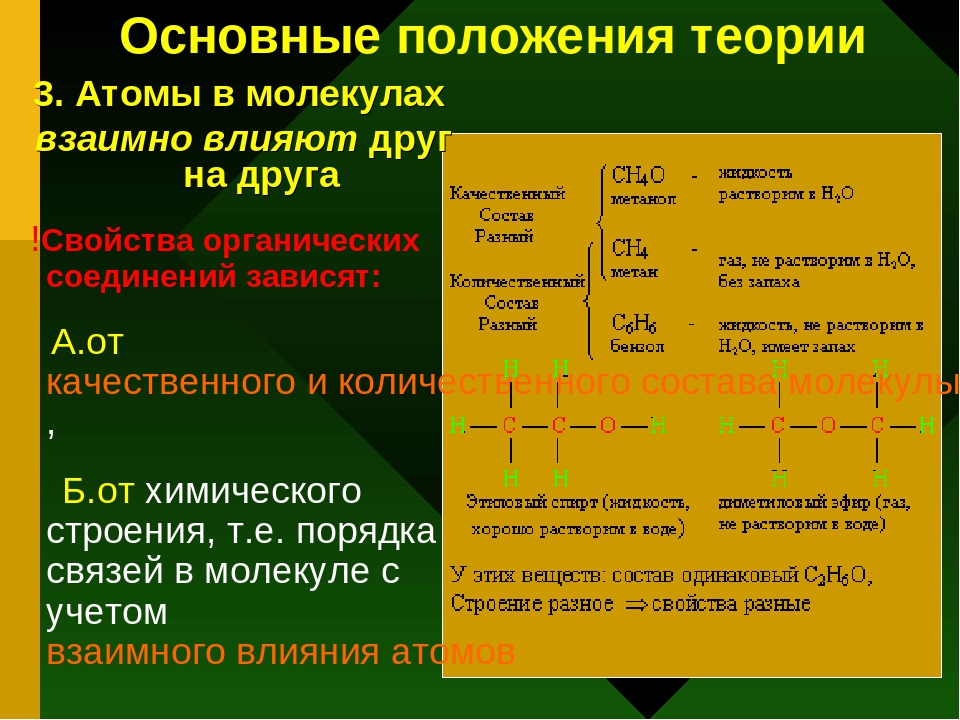
В 1869 г. И.Вислиценус (1835-1902), пытаясь понять случаи изомерии, которые не находили объяснения в классической теории химического строения, например малеиновой и фумаровой кислот, высказал идею о пространственном расположении атомов в молекулах и ввел понятие «геометрическая изомерии», т.е. цис-, транс-изомерия для производных этилена:
В 1874 г. идею И.Вислиценуса развили
независимо друг от друга голландец
Я.Вант-Гофф (1852-1911) и француз
Ж.Ле Бель (1847-1930). Они исходили
из того, что формулы соединений
должны выражать пространственное
строение органических молекул. Так, в
молекуле метана и его производных
атом углерода должен располагаться в
центре тетраэдра, а четыре
заместителя должны быть помещены в
вершины тетраэдра. В тех случаях,
когда с атомом углерода связаны
четыре различных заместителя, атом
углерода становится асимметрическим
и химическое соединение становится
оптически активным. И снова против
этих идей резко выступил в 1877 г.
Г.Колбе в статье «Приметы времени».
М.Бертло также в принципе отверг
возможность установления строения
сложных веществ. Еще и сейчас в
учебной литературе структурные
формулы считаются как бы
неполноценными, не выражающими
пространственного строения молекул. В
заключение мы приводим фрагмент
структурной формулы молекулы ДНК, в
которой зашифрована вся
генетическая информация человека
(молекулы дезоксирибонуклеиновой
кислоты). Классическая теория
химического строения прекрасно
описывает стуктуру этой сложной
молекулы.
В тех случаях,
когда с атомом углерода связаны
четыре различных заместителя, атом
углерода становится асимметрическим
и химическое соединение становится
оптически активным. И снова против
этих идей резко выступил в 1877 г.
Г.Колбе в статье «Приметы времени».
М.Бертло также в принципе отверг
возможность установления строения
сложных веществ. Еще и сейчас в
учебной литературе структурные
формулы считаются как бы
неполноценными, не выражающими
пространственного строения молекул. В
заключение мы приводим фрагмент
структурной формулы молекулы ДНК, в
которой зашифрована вся
генетическая информация человека
(молекулы дезоксирибонуклеиновой
кислоты). Классическая теория
химического строения прекрасно
описывает стуктуру этой сложной
молекулы.
В настоящее время классическая
теория химического строения получила
дальнейшее развитие в работах
профессора МГУ В. М.Татевского, его
учеников, и в рамках компьютерной
химии (в работах академика
Н.С.Зефирова).
М.Татевского, его
учеников, и в рамках компьютерной
химии (в работах академика
Н.С.Зефирова).
см. также
Дальнейшее развитие теории химического строения
Теория химического строения А. М. Бутлерова. … Дальнейшее развитие теории химического строения. Борьба [c.310]Одна из очередных задач дальнейшего развития теории химического строения [c.56]
Таким образом, при помощи метода молекулярных орбиталей успешно объясняют свойства различных молекул. Этот метод важен тем, что позволяет получить данные о свойствах молекул исходя из соответствующих характеристик атомов. Метод МО не исключает метода валентных связей, оба метода взаимно дополняют друг друга. В целом оба метода (и ВС, и МО) служат квантовомеханическим обоснованием и дальнейшим развитием теории химического строения А. М. Бутлерова. [c.62]
В структурных формулах различная сила химической связи отдельных атомов изображалась одинаково.
 Дальнейшее развитие теории химического строения выявило несоответствие между содержанием понятия структура и формой ее выражения. [c.195]
Дальнейшее развитие теории химического строения выявило несоответствие между содержанием понятия структура и формой ее выражения. [c.195] По вопросу дальнейшего развития теории химического строения Бутлеров писал Само собой разумеется, что, когда мы будем знать ближе натуру химической энергии, самый род атомного движения,— когда законы механики получат и здесь приложение,— тогда учение о химическом строении падет, как падали прежние химические теории, но, подобно большинству этих теорий, оно падет не для того, чтобы исчезнуть, а для того, чтобы войти в измененном виде в круг новых и более широких воззрений . Итак, автор теории химического строения предвидел приложение механики атом-ного мира (т. е. квантовой механики) к его теории. Именно применение квантовой механики к проблемам структуры вещества подняло теорию химического строения Бутлерова на новую, высшую ступень. Только в одном не прав был Бутлеров его теория не пала, а превратилась в общехимическую теорию, являющуюся фундаментом современной химии. [c.12]
[c.12]
ДАЛЬНЕЙШЕЕ РАЗВИТИЕ ТЕОРИИ ХИМИЧЕСКОГО СТРОЕНИЯ. [c.143]
Появление новых направлений исследований связи между строением и запахом органических соединений было обусловлена дальнейшим развитием теории химического строения — возникновением стереохимии. [c.128]
Для дальнейшего развития теории химического строения большое значение получило учение о взаимном влиянии атомов в молекулах соединений. Сама идея взаимного влияния атомов уже фигурировала в докладе А. М. Бутлерова (1861). В даль- нейшем (1863) он указал, что многоатомные элементы проявляют в сложных молекулах различные отношения в зависимости от природы элементов, с которь ми они связаны. Так, атом водорода ведет себя различно, соединен ли он с углеродом или с кислородом. [c.146]
В последние два-три десятилетия был получен большой экспериментальный материал, который потребовал дальнейшего развития теории химического строения. Основная масса новых фактов относится к проблеме взаимного влияния атомов в молекулах. Важнейшие из этих фактов связаны с взаимным влиянием непосредственно не связанных атомов, передачей взаимного влияния по цепи сопряженных связей, нарушением взаимного влияния вследствие пространственных препятствий, реакционной способностью ароматических и диеновых соединений, а также с устойчивостью некоторых свободных радикалов, цветностью органических соединений и с многими другими явлениями. [c.50]
Основная масса новых фактов относится к проблеме взаимного влияния атомов в молекулах. Важнейшие из этих фактов связаны с взаимным влиянием непосредственно не связанных атомов, передачей взаимного влияния по цепи сопряженных связей, нарушением взаимного влияния вследствие пространственных препятствий, реакционной способностью ароматических и диеновых соединений, а также с устойчивостью некоторых свободных радикалов, цветностью органических соединений и с многими другими явлениями. [c.50]
В дальнейшем развитии теории химического строения оказалось, что среди тех отношений атомов в молекуле, которые были определены выше как отношения непосредственной химической зависимости (отношения химической связи), имеются резкие качественные и количественные градации. Именно, химические связи, осуществляющиеся в различных соединениях между многовалентными атомами, могут быть различной кратности, т. е. на образование связи между многовалентными атомами последние могут затрачивать не только по одной, но также по две или три единицы сродства .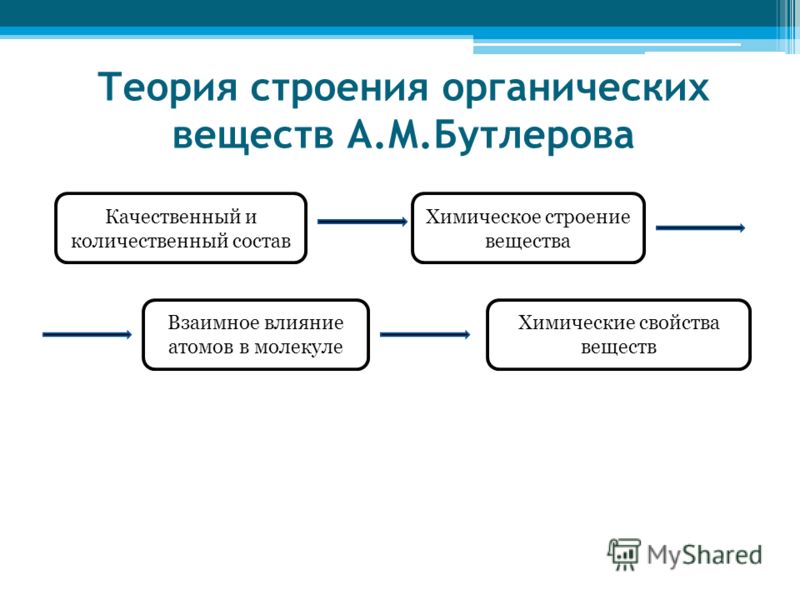 Таким образом, возникло представление об ординарных, двойных и тройных (и вообще кратных ) связях. [c.30]
Таким образом, возникло представление об ординарных, двойных и тройных (и вообще кратных ) связях. [c.30]
В этом высказывании А. М. Бутлерова ясно проводится та мысль, что одно из важнейших направлений дальнейшего развития теории химического строения должно состоять в детальном исследовании молекулы как динамической, подвижной, способной к изменению системы, в детальном исследовании внутренних составных частей, внутренних движений в молекуле, в установлении связей, закономерностей между [c.36]
Следует особо отметить, что при этом дальнейшем развитии теории химического строения ее основные положения, сформулированные Бутлеровым, остались незыблемыми. [c.47]
ПУТИ ДАЛЬНЕЙШЕГО РАЗВИТИЯ ТЕОРИИ ХИМИЧЕСКОГО СТРОЕНИЯ А. М. БУТЛЕРОВА [c.49]
Дальнейшее развитие теории химического строения (стереохимическая теория) [c.84]
Дальнейшее развитие теории химического Строения было связано с проблемой установления и объяснения изомерии. Э. Эрленмейер был почти единственным ученым, с самого начала принявшим главные идеи теории химического строения, высказанные в докладе А. М. Бутлерова (1861). С этого времени между учеными завязалась переписка и установились дружеские отношения. А. М Бутлеров опубликовал многие свои статьи по теории химического строения в журнале, издававшемся Э. Эрленмейером Но в целом теорию химического строения Э. Эрленмейер принял не сразу. Еще в 1863 г. он полагал, например, что диметил (этан) СНз-СНз и так называемый водородистый этил aHs-H представляют собой различные соединения. В связи с этим он считал, что четыре единицы валентности углерода неодинаковы. Это допущение приводило к неправильному выводу, что число изомеров различных соединений должно быть значительно расширено. Однако Э. Эрленмейер отказался от своих утверждений лишь после того, как К. Шорлеммер [c.144]
Э. Эрленмейер был почти единственным ученым, с самого начала принявшим главные идеи теории химического строения, высказанные в докладе А. М. Бутлерова (1861). С этого времени между учеными завязалась переписка и установились дружеские отношения. А. М Бутлеров опубликовал многие свои статьи по теории химического строения в журнале, издававшемся Э. Эрленмейером Но в целом теорию химического строения Э. Эрленмейер принял не сразу. Еще в 1863 г. он полагал, например, что диметил (этан) СНз-СНз и так называемый водородистый этил aHs-H представляют собой различные соединения. В связи с этим он считал, что четыре единицы валентности углерода неодинаковы. Это допущение приводило к неправильному выводу, что число изомеров различных соединений должно быть значительно расширено. Однако Э. Эрленмейер отказался от своих утверждений лишь после того, как К. Шорлеммер [c.144]
Важным направлением исследований, связанных с дальнейшим развитием теории химического строения, была разработка проблемы взаимного влияния атомов в химических соединениях. Эта проблема была широко изучена и проанализирована в докторской диссертации Марковникова Материалы к вопросу о взаимном влиянии атомов в химических соединениях (1869). В этой работе проблема взаимного влияния атомов рассматривалась как неотъемлемая и существенная часть теории химического строения. Объясняя природу взаимного влияния атомов, Марковников привлекает представления о химическом сродстве и утверждает, что именно проявлением сил химического сродства обусловлено взаимное влияние атомов. В этом отношении он целиком следует представлениям Бутлерова. [c.316]
Эта проблема была широко изучена и проанализирована в докторской диссертации Марковникова Материалы к вопросу о взаимном влиянии атомов в химических соединениях (1869). В этой работе проблема взаимного влияния атомов рассматривалась как неотъемлемая и существенная часть теории химического строения. Объясняя природу взаимного влияния атомов, Марковников привлекает представления о химическом сродстве и утверждает, что именно проявлением сил химического сродства обусловлено взаимное влияние атомов. В этом отношении он целиком следует представлениям Бутлерова. [c.316]
Большим и важным событием в дальнейшем развитии теории химического строения явилось установление химического строения бензола и ароматических соединений. [c.317]
Теория замещения могла бы устоять только с падением теории химического строения. Меншуткин собрал все доводы, чтобы опрокинуть последнюю. Его критика, иногда неумеренно пристрастная иногда направленная на мелочи, имела одну положительную сторону — она подняла почти все вопросы, стоявшие в порядке дня и требовавшие своего разрешения для дальнейшего развития теории химического строения. [c.255]
[c.255]
Большое значение для дальнейшего развития теории химического строения получило учение о взаимном влиянии атомов в молекулах соединений. Сама идея взаимного влияния атомов в молекулах фигурировала уже в докладе Бутлерова в 1861 г. В дальнейшем эта идея получила в исследованиях Бутлерова определенное развитие. Бутлеров указал (1863), что атомы в молекулах проявляют различные свойства в зависимости от природы элементов, с которыми они соединены. Так, атом водорода ведет себя совер-гпенно различно (по отношению к реагентам), в зависимости от того, соединен он с углеродом или кислородом [c.314]
А. М. Зайцев поставил перед собою новую не менее важную задачу в области изомерии амиловых спиртов, решение которой, несомненно, должно было способствовать дальнейшему развитию теории химического строения вещества. [c.26]
Открытие электронов и появление теории Бора (1913) строения атома способствовали дальнейшему развитию теории химического строения, так как в органическую химию были введены электронные представления. Одну из первых электронных гипотез в органической химии выдвинул в России в 1914—1916 г. А. М. Беркенгейм. В основе его взглядов лежали электростатические представления. Согласно гипотезе А. М. Беркеигейма, углерод выполняет смешанные функции, являясь электроположительным по отношению к таким атомам, как хлор, и электроотрицательным по отношению к таким атомам, как водород. Эти смешанные функции атом углерода выполняет одновременно по отношению к разным атомам, с которыми он связан. Согласно теории А. М. Беркеигейма, химическая связь осуществляется одним электроном. В неорганических соединениях этот электрон может пол—ностью перейти от одного атома к другому, после чего атомы будут удерживаться только электростатическими силами. В органических молекулах валентный электрон полностью не переходит от одного атома к другому, а только смещается по направлению к одному из них, например, от углерода к хлору, от водорода к углероду и т. п.. [c.29]
Одну из первых электронных гипотез в органической химии выдвинул в России в 1914—1916 г. А. М. Беркенгейм. В основе его взглядов лежали электростатические представления. Согласно гипотезе А. М. Беркеигейма, углерод выполняет смешанные функции, являясь электроположительным по отношению к таким атомам, как хлор, и электроотрицательным по отношению к таким атомам, как водород. Эти смешанные функции атом углерода выполняет одновременно по отношению к разным атомам, с которыми он связан. Согласно теории А. М. Беркеигейма, химическая связь осуществляется одним электроном. В неорганических соединениях этот электрон может пол—ностью перейти от одного атома к другому, после чего атомы будут удерживаться только электростатическими силами. В органических молекулах валентный электрон полностью не переходит от одного атома к другому, а только смещается по направлению к одному из них, например, от углерода к хлору, от водорода к углероду и т. п.. [c.29]
В связи с прошедшими дискуссиями, показавшими научную песочу стоятельность, идеалистическую сущность теории резонанса Паулинга и теории мезомерии Ингольда, особенно важное значение приобретает задача дальнейшего развития теории химического строения А. М. Бут-лерова. [c.17]
М. Бут-лерова. [c.17]
А. М. Бутлеров говоэит следующее о путях дальнейшего развития теории химического строения Само собою разумеется, что когда мы будем знать ближе натуру химической энергии, самый род атомного движения, — когда законы механики получат и здесь приложение, тогда учение о химическом строении падет, как падали прежние химические теории, но, подобно большинству этих теорий, оно падет не для того, чтобы исчезнуть, для того, чтобы войти в измененном виде в круг новых и более широких воззрений ([2], стр. 431—432). [c.36]
Открытие электронов, установление того факта, что электроны входят в состав каладого атома, и появление теории Бора (1913) о строении атома способствовали дальнейшему развитию теории химического строения благодаря введению в органическую химию электронных представлений. [c.96]
Приведенные мною примеры указывают на плодотворность применения метода изучения электронной плотности к проблеме исследования природы химической связи. Представление строения молекул органических соединений, так же как и других объектов, в виде распределения электронной плотности позволяет рассматривать молекулу как единое целое в соответствии с теорией химического строения Бутлерова, который говорил Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определенным количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которого химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу (А. М. Бутлеров. Избр. работы по органической химии. Изд. АН СССР, 1951, стр. 71—72). Согласно современным представлениям, химические силы обусловлены валентными электронами атомов, и, следовательно, изучение распределения электронной плотности является основной задачей современного развития теории химического строения как органических, так и других соединений.
Представление строения молекул органических соединений, так же как и других объектов, в виде распределения электронной плотности позволяет рассматривать молекулу как единое целое в соответствии с теорией химического строения Бутлерова, который говорил Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определенным количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которого химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу (А. М. Бутлеров. Избр. работы по органической химии. Изд. АН СССР, 1951, стр. 71—72). Согласно современным представлениям, химические силы обусловлены валентными электронами атомов, и, следовательно, изучение распределения электронной плотности является основной задачей современного развития теории химического строения как органических, так и других соединений. В настоящее время еще очень мало внимания уделяется прямому определению (при помощи эксперимента) распределения электронной плотности. Работа эта в экспериментальном отношении очень сложная и требует значительных усилий от исследователя, но большое значение полученных этим методом результатов требует значительного расширения работ по определению электронной плотностн. Совокупное применение синтеза, изучения химических и физических свойств и определения электронной плотности, несомненно, будет способствовать дальнейшему развитию теории химического строения Бутлерова. [c.196]
В настоящее время еще очень мало внимания уделяется прямому определению (при помощи эксперимента) распределения электронной плотности. Работа эта в экспериментальном отношении очень сложная и требует значительных усилий от исследователя, но большое значение полученных этим методом результатов требует значительного расширения работ по определению электронной плотностн. Совокупное применение синтеза, изучения химических и физических свойств и определения электронной плотности, несомненно, будет способствовать дальнейшему развитию теории химического строения Бутлерова. [c.196]
Дальнейшее развитие теории химического строения А. М. Бутлерова требует, естественно, использования современных электронных нредстав лений о строении вещества. Использование электронных представлений должно развиваться и сопровождаться точным и строгим анализом того, что подразумевается под каждым понятием, под каждым эффектом, отбрасывая все идеалистическо-махистское. В этом плане необходимо провести большую и кропотливую работу и подготовить ко. плективную монографию о современных представлениях в органической химии. [c.411]
плективную монографию о современных представлениях в органической химии. [c.411]
Особенно быстрый прогресс современной химии был вызван проникновением в эту науку представлений об электронном строении атомных и молекулярных оболочек. Не случайно дальнейшее развитие теории химического строения А. М. Бутлерова и положений В. В. Мар-ковникова о взаимном влиянии атомов в молекуле связано с установлением электростатического характера химической связи, а затем — ее электронной природы. Химическая связь —центральная, главнейшая проблема химии. Поэтому некоторые химики справедливо полагают, что раскрыть природу, характер химической связи, закономерности образования и разрушения ее —значит раскрыть механизм и существо процесса превращения веществ, т. е. самую суть химического движения. [c.40]
Основные положения теории химического строения органических веществ
Содержание:
Основные положения теории химического строения органических веществ
- Основные положения теории химического строения органических веществ Создание теории строения органических соединений связано с несколькими именами химиков 19 века.
 К. Франкленд, С. Жерар, А. Кукул, А. Купер и др. Решающая роль в развитии этой теории (1861) принадлежит великому русскому ученому Александру Михайловичу буторову.
К. Франкленд, С. Жерар, А. Кукул, А. Купер и др. Решающая роль в развитии этой теории (1861) принадлежит великому русскому ученому Александру Михайловичу буторову.
 К. Франкленд, С. Жерар, А. Кукул, А. Купер и др. Решающая роль в развитии этой теории (1861) принадлежит великому русскому ученому Александру Михайловичу буторову.
К. Франкленд, С. Жерар, А. Кукул, А. Купер и др. Решающая роль в развитии этой теории (1861) принадлежит великому русскому ученому Александру Михайловичу буторову.- Он ввел понятие химического строения вещества(понимал порядок связей атомов в молекуле), установил, что химическое строение вещества определяет его свойства, и показал, что его теория имеет большое практическое значение в предсказании новых веществ. А. М. Бутлеров (1828-1886) сформулировал основную идею теории строения органических веществ.
1.Все атомы, образующие молекулы органических веществ, соединены в определенном порядке в соответствии с их валентностью. Людмила Фирмаль
Порядок связей атомов в молекуле и характер их связей А. М. Баттереров называет химическим structure. In в соответствии с этими выражениями валентность элементов представлена условно в тире. Например, структура молекулы метана может быть показана следующим образом: Н. я. Х. Я Н Принципиальная схема такой молекулярной структуры называется структурной или структурной формулой.
я. Х. Я Н Принципиальная схема такой молекулярной структуры называется структурной или структурной формулой.
Исходя из положений о свойствах четырехвалентного углерода и способности его атомов образовывать цепи и кольца, структурную формулу органических веществ можно выразить следующим образом: Н. Н. Н. Я s2n6 н-ы-ы-н Этан| Дж Н. Н. Н. Н. Н. Н. Я. s3n8•н-ы-ы-ы-н Пропан Н. Н. Н. Н2С-СН2 C5H, 0 I I Циклопентан Пи вопрос 2khtsn / 2.Свойства вещества зависят не только от атома и числа, содержащихся в составе молекулы, но и от порядка атомов в ней.
Такое положение теории строения органических веществ, в частности, объясняло явление изомерии. Существуют соединения, которые содержат одинаковое количество атомов одного и того же элемента, но соединены в разном порядке. Такие соединения обладают различными свойствами и называются isomers.
- So например, 2 вещества соответствуют одной и той же молекулярной формуле C4H, 0. Н Н — Н-Н-Н-К-Н Н-н-н-н — н-н-н-н-н-н. j’J Изобутан (температура кипения-11,7°C) Изомерия-это явление, при котором существует несколько веществ с одинаковым составом и одинаковой молекулярной массой, но с разными молекулярными структурами.
 j’J Изобутан (температура кипения-11,7°C) Изомерия-это явление, при котором существует несколько веществ с одинаковым составом и одинаковой молекулярной массой, но с разными молекулярными структурами.
j’J Изобутан (температура кипения-11,7°C) Изомерия-это явление, при котором существует несколько веществ с одинаковым составом и одинаковой молекулярной массой, но с разными молекулярными структурами.Органическая химия очень распространена. Познакомьтесь с ней при изучении органических веществ во всех классах. 3.В зависимости от свойств данного вещества можно определить структуру его молекулы и предсказать ее свойства по структуре молекул.
Например, свойства неорганических веществ зависят от структуры кристаллической решетки (I, с. 130). Людмила Фирмаль
Характерные свойства атомов от ионов объясняются их структурой (s: 5). в дальнейшем молекулярная формула будет одинаковой, а структура будет подтверждать различные физические и химические свойства различных органических веществ. 4.Атомы и группы атомов в молекуле вещества взаимно влияют друг на друга other. As вы уже знаете, что свойства неорганических соединений с гидроксо группой зависят от атомов, которые они объединяют-металлов или non-metals.
So например, гидроксо-группа включает в себя как основания, так и кислоты. Ч- ° вп Н3-0-з-н-пр) Однако свойства этих веществ совершенно различны. В будущем мы убедимся, что органические соединения также обладают различными свойствами. Эти свойства зависят от атомов или групп атомов, с которыми связана гидроксильная группа. Вопрос 8-12 (стр. 147).
Смотрите также:
Решение задач по химии
Теория химического строения А .М. Бутлерова.для 9 класса
Тема4 урок№2: Теория химического строения А.М. Бутлерова.
Следовать за мыслями
великого человека
есть наука самая
замечательная.
А.С.Пушкин
Смысл нельзя дать,
его нужно найти
В.Франкл
Задачи:
Образовательные:
формировать понятия о сущности теории химического строения органических веществ, опираясь на знания учащихся об электронном строении атомов элементов, их положении в Периодической системе Д. И. Менделеева, о степени окисления, природе химической связи и о других главнейших теоретических положениях:
И. Менделеева, о степени окисления, природе химической связи и о других главнейших теоретических положениях:
последовательность расположения атомов углерода в цепи,
взаимное влияние атомов в молекуле,
зависимость свойств органических веществ от структуры молекул;
сформировать представление о ходе развития теорий в органической химии;
усвоить понятия: изомеры и изомерия;
разъяснить смысл структурных формул орг.веществ и их преимуществ перед молекулярными;
показать необходимость и предпосылки создания теории химического строения;
продолжить формирование навыков составления конспекта.
Развивающие:
развивать мыслительные приемы анализа, сравнения, обобщения;
развивать абстрактное мышление;
тренировать внимание учащихся при восприятии большого по объему материала;
выробатывать умения анализировать информацию и выделять наиболее важный материал.
Воспитательные:
с целью патриотического и интернационального воспитания привести учащимся
исторические сведения о жизни и деятельности ученых
Оборудование: портрет А. М.Бутлерова, карточки – схемы, стенд о А.М. Бутлерове, модели атомов, книга А.Ю. Алекберовой.
М.Бутлерова, карточки – схемы, стенд о А.М. Бутлерове, модели атомов, книга А.Ю. Алекберовой.
Ход урока
I. Актуализация опорных знаний.
Из письма Вёлера Берцелиусу (1835)
«Органическая химия может сейчас кого угодно свести с ума. Она представляется мне дремучим лесом, полных удивительных вещей, безграничной чащей, из которой нельзя выбраться, куда не осмеливаешься проникнуть»
Мир сложен.
Он полон событий, сомнений,
И тайн бесконечных, и смелых догадок…
Как чудо природы рождается гений
И в хаосе этом находит порядок.
Фронтальная беседа
1. Кто и когда ввёл понятие «органическая химия»? Какие вещества в то время назывались органическими?
2. Какие вещества в то время назывались органическими?
3. Какие синтезы подтолкнули к развитию органическую химию?
4. Назовите учёных и их открытия, которые явились научной базой для создания теории химического строения органических веществ. Дайте определение понятию «валентность».
Дайте определение понятию «валентность».
5. Валентность в органических веществах углерода, водорода, кислорода, азота.
II. Изучение нового материала
1. Введение в тему урока.
Учитель:
Окружающий нас мир гармоничен, но не все могут прочувствовать это. В природе, музыке, искусстве, в науке присутствует своя гармония. И лишь немногие очень талантливые люди способны донести звучание этой гармонии до большинства.
Гением в неорганической химии, нашедшем гармонию химических элементов явился Д.И.Менделеев, создавший свой великий закон.
Сегодня мы с вами прислушаемся к словам одного великого человека, чьи слова прозвучали в эпиграфе и последуем за мыслями другого великого человека – основателя теории химического строения А.М.Бутлерова, которая определила присутствие гармонии в науке – органической химии.
Целью нашего урока является повторить, углубить, рассмотреть основные положения теории о строении органических веществ на более высоком качественном уровне, с позиции имеющихся знаний об органических веществах . При этом совместную работу мы постараемся организовать так, чтобы во всём вы сами искали смысл.
При этом совместную работу мы постараемся организовать так, чтобы во всём вы сами искали смысл.
2.Необходимость теоретического обоснования
а) имперический период развития химии
И. Берцелиус – середина XVII – конец XVIII вв – орг. химия – растительные и животные вещества.
Парацельс – искал лекарственные препараты
б) аналитический период
( конец XVIII – середина XIX вв) изучение состава веществ
в) органический синтез
До начала 19-века все известные вещества делили по их происхождению на две группы: вещества минеральные и вещества органические. Многие ученые тех времен считали, что органические вещества могут образоваться только в живых организмах при помощи «жизненной силы». Такие взгляды назывались виталистическими. Большой удар взглядам виталистов нанес немецкий химик Ф. Велер. Он первые получил органические вещества из неорганических: в 1824 г. – щавелевую кислоту, а в 1828 г. – мочевину. Щ. к. встречается в растениях, а мочевина образуется в организме человека и животных.
– мочевину. Щ. к. встречается в растениях, а мочевина образуется в организме человека и животных.
Большинство неорганических веществ немолекулярного строения, и поэтому они обладают высокими температурами плавления и кипения. Органические вещества, как правило, молекулярного строения, и поэтому они имеют низкие температуры плавления. В состав молекул всех органических веществ входит углерод. Органическая химия – это раздел химической науки, в котором изучаются соединения углерода и их превращения. Органические вещества – углеродосодержащие вещества, которые образуются в живых организмах, которые синтезируются.
3. История становления теории химического строения
1. Первые попытки создания теории.
а) Теория радикалов
С древнейших времен человечеству известны различные соединения углерода растительного и животного происхождения и некоторые способы их получения и переработки.
Например:
– из цветов извлекали эфирные масла;
– нагревая жир с содой, получали мыло;
– сбраживая виноградный сок, получали вино, а при его перегонке — спирт;
– в Древней Индии, Финикии, Египте для крашения использовали растительные красители — пурпур, индиго, ализарин.
Однако в тот период, вплоть до начала XIX в., не делали различия между органическими и неорганическими веществами.
В 1807 г. известный уже вам шведский ученый И.Я. Берцелиус предложил выделить изучение веществ растительного и животного происхождения в самостоятельную дисциплину — органическую химию. И.Я. Берцелиусасчитают родоначальником органической химии.
Но становление органической химии как науки произошло во второй половине XIX века. Химики того времени делали попытки понять особенности строения и химического поведения органических веществ, синтезировать органические соединения. Среди этих попыток были и успешные.
30 –е годы XIX в И.Я. Берцелиус, Ю Либих, Ф. Вёлер
Они полагали, что органические вещества построены из двух частей -радикалов. Он считал, что радикалы сохраняются неизменными во время химических реакций.
Теория радикалов была постепенно отвергнута, однако она оставила глубокий след в органической химии
– понятие «радикал» прочно вошло в химию;
– верным оказалось утверждение о возможности существования радикалов в свободном виде, о переходе в огромном числе реакций определенных групп атомов из одного соединения в другое.
Недостаток: была формальна
б) Теория типов
40 – 50 гг XIX в Г.Ш.Жерар, Ф.А.Кекуле
Предложили рассматривать каждое вещество как единое целое. Они считали, что все органические соединения являются производными простейших неорганических веществ: водорода, хлористого водорода, воды и предложил несколько типов.
I тип — водорода II тип – хлористого водорода
III тип – воды
Теория типов сыграла большую роль в развитии органической химии
– позволила предсказать и открыть ряд веществ;
– оказала положительное влияние на развитие учения о валентности;
– обратила внимание на изучение химических превращений органических соединений, что позволило глубже изучить свойства веществ, а также свойства предсказываемых соединений;
– создала совершенную для того времени систематизацию органических соединений.
Недостаток: для некоторых веществ можно было составить несколько разных формул.
4. Предпосылки появления ТХС
а) Важным этапом был съезд химиков в Карлсруэ в 1860 году, где были развиты и подтверждены идеи атомистики.
б) В результате работ Э. Франкланда и Кекуле в химии утвердилось понятие о валентности, и, в частности, было развито представление о 4-валентности углерода (Кекуле).
в) А в 1857 году Ф. А. Кекуле и А. С. Купер выдвинули идею о соединении атомов углерода в цепи.
Вывод: все эти успехи науки подготовили условия для нового этапа в развитии органической химии — появление теории химического строения органических соединений.
5. Основные идеи и положения теории
а)природа вещества
химическая натура сложной частицы определяется натурой элементарных составных частиц, количеством их и химическим строением
Для молекул важно не только количество атомов, но и порядок их соединения » т.е. строение. Сейчас это очевидно, а где-то 170 – 180 лет назад химики этому значения не придавали. И только ТХС А.М.Бутлерова помогла.
И только ТХС А.М.Бутлерова помогла.
б) основные положения
И так, к середине 19-го века было создано немало теорий, авторы которых пытались объяснить эти и другие особенности органических соединений. Одной из таких теорий стала теория химического строения Бутлерова*.
* Бутлеров Александр Михайлович (15.09.1928–17.08.1886) — русский химик. Создал теорию химического строения органических веществ, лежащей в основе современной химии. Предсказал изомерию многих органических соединений, заложил основы учения о таутомерии. (от греч. ταύτος — тот же самый и μέρος — мера) — явление обратимой изомерии, при которой два или более изомера легко переходят друг в друга.[1] При этом устанавливается таутомерное равновесие, и вещество одновременно содержит молекулы всех изомеров (таутомеров) в определённом соотношении.
Некоторые её положения были изложены А. М. Бутлеровым в 1861 году на конференции в г. Шпейере, другие были сформулированы позже в научных работах А. М. Бутлерова. В целом, основные положения этой теории в современном изложении можно сформулировать так.
Шпейере, другие были сформулированы позже в научных работах А. М. Бутлерова. В целом, основные положения этой теории в современном изложении можно сформулировать так.
1. Атомы в молекулах располагаются в строгом порядке, согласно их валентности.
2. Атом углерода в органических молекулах всегда имеет валентность равную четырём.
3. Порядок соединений атомов в молекуле и характер химических связей между атомами называется химическим строением.
4. Свойства органических соединений зависят не только от того, какие атомы и в каких количествах входят в состав молекулы, но и от химического строения:
вещества разного строения имеют разные свойства;
вещества похожего строения имеют похожие свойства.
5. Изучая свойства органических соединений, можно сделать вывод о строении данного вещества и описать это строение одной-единственной химической формулой.
6. Атомы в молекуле влияют друг на друга, и это влияние сказывается на свойствах вещества.
6. значение теории химического строения
Создание теории строения веществ сыграло важнейшую роль в развитии органической химии.:
1. Из науки преимущественно описательной она превращается в науку созидательную, синтезирующую, появилась возможность судить о взаимном влиянии атомов в молекулах различных веществ.
2. Теория строения создала предпосылки для объяснения и прогнозирования различных видов изомерии органических молекул, а также направлений и механизмов протекания химических реакций.
3. На основе этой теории химики-органики создают вещества, которые не только заменяют природные, но по своим свойствам значительно их превосходят. Так, синтетические красители гораздо лучше и дешевле многих природных, например известных в древности ализарина и индиго. В больших количествах производят синтетические каучуки с самыми разнообразными свойствами. Широкое применение находят пластмассы и волокна, изделия из которых используют в технике, быту, медицине, сельском хозяйстве.
В больших количествах производят синтетические каучуки с самыми разнообразными свойствами. Широкое применение находят пластмассы и волокна, изделия из которых используют в технике, быту, медицине, сельском хозяйстве.
А.М.Бутлеров сам доказал справедливость своей теории, предсказав существование двух изомеров составаС4Н10 и синтезировав изобутан.
Итак, теория строенияобъяснила неясности и противоречия в знаниях об органических веществах,определила качественно новый подход к пониманию строения соединений,открыла путь для синтеза новых органических соединений.
Значение теории
объяснила неясности и противоречия в знаниях об органических веществах;
творчески обобщила достижения в области химии;
определила качественно новый подход к пониманию строения соединений;
стала основой для объяснения и прогнозирования свойств органических веществ;
открыла путь для синтеза новых органических соединений.

Отстаивая свое учение о химическом строении и показывая его практическую значимость, А.М. Бутлеров не считал это учение абсолютным и неизменным. Действительно, если молекула — это реальность, построенная из реальных атомов, то она должна представлять собой определенное физическое тело в трехмерном пространстве.
III.Контроль знаний
Ещё в древности Демокрит сказал: «Суть дела не в полноте знания, а в полноте разумения». Поэтому я предлагаю вам, используя полученные знания выполнить несколько заданий
Задание 1: Составьте структурные формулы двух изомеров (одинаковый состав, но разный порядок соединения атомов) состава С4Н10, расположив их друг напротив друга.
Задание 2: Выпишите: Изомеры состава С5Н12 – 1 вариант;
Изомеры состава С6Н14 – 2 вариант;
СН3-СН2-СН2-СН2-СН2-СН3, 2) СН3-СН-СН2-СН3 ,
СН3
3) СН3-СН2-СН2-СН-СН3, 4) СН3-СН2-СН2-СН2-СН3,
СН3
5)СН3-СН-СН3, 6)СН3-СН2-СН2-СН3׀
СН3
IV. Рефлексия
Что нового узнали сегодня на уроке?
Что в материале показалось трудным?
С чем справились легко?
Полезен ли материал сегодняшнего урока?
V. Подведение итогов урока
Отметки за урок выставляем совместно.
VI..Домашнее задание: § 35
Бутлеров Александр Михайлович. Разработка теории химического строения органических соединений
Александр Михайлович Бутлеров родился 3 (15) сентября 1828 года в городе Чистополь Казанской губернии в семье помещика, офицера в отставке. Первое образование получил в частном пансионе, затем учился в гимназии и Казанском императорском университете. С 1849-го преподавал, в 1857-м стал ординарным профессором химии в том же университете. Дважды был его ректором. В 1851-м защитил магистерскую диссертацию «Об окислении органических соединений», а в 1854-м в Московском университете — докторскую диссертацию «Об эфирных маслах». С 1868 года был ординарным профессором химии Петербургского университета, с 1874-го — ординарным академиком Петербургской академии наук. Кроме химии Бутлеров уделял внимание практическим вопросам сельского хозяйства, садоводству, пчеловодству, под его руководством началось разведение чая на Кавказе. Умер в деревне Бутлеровка Казанской губернии 5 (17) августа 1886 года.
До Бутлерова предпринималось немалое количество попыток создать учение о химическом строении органических соединений. К этому вопросу не раз обращались самые именитые химики того времени, работы которых частично были использованы русским ученым для своей теории строения. Например, немецкий химик Август Кекуле пришел к выводу, что углерод может образовывать четыре связи с другими атомами. Более того, он считал, что для одного и того же соединения может существовать несколько формул, однако при этом всегда добавлял, что в зависимости от химического превращения эта формула может быть разной. Кекуле полагал, что формулы не отражают того, в какой последовательности соединены атомы в молекуле. Другой видный немецкий ученый, Адольф Кольбе, вообще считал принципиально невозможным выяснение химического строения молекул.
Свои основные идеи о строении органических соединений Бутлеров впервые высказал в 1861 году в докладе «О химическом строении вещества», который представил на суд участников Съезда немецких естествоиспытателей и врачей в Шпейере. В свою теорию он включил идеи Кекуле о валентности (количестве связей для конкретного атома) и шотландского химика Арчибальда Купера о том, что атомы углерода могут образовывать цепочки. Принципиальным отличием теории Бутлерова от других было положение о химическом (а не механическом) строении молекул — способе, с помощью которого атомы связывались друг с другом, образовывая молекулу. При этом каждый атом устанавливал связь в соответствии с принадлежащей конкретно ему «химической силой». В своей теории ученый проводил четкое различие между свободным атомом и атомом, вступившим в соединение с другим (он переходит в новую форму, а в результате взаимного влияния соединенные атомы, в зависимости от структурного окружения, имеют различные химические функции). Русский химик был убежден, что формулы не просто схематично изображают молекулы, но и отражают их реальное строение. Более того, каждая молекула имеет определенную структуру, которая меняется только в ходе химических превращений. Из положений теории следовало (впоследствии было подтверждено экспериментально), что химические свойства органического соединения определяются его строением. Это утверждение особенно важно, так как позволило объяснять и предсказывать химические превращения веществ. Существует и обратная зависимость: по структурной формуле можно судить о химических и физических свойствах вещества. Кроме этого, ученый обратил внимание на то, что реакционная способность соединений объясняется энергией, с которой связываются атомы.
С помощью созданной теории Бутлеров смог объяснить изомерию. Изомерами называют соединения, количество и «качество» атомов в которых одинаково, но при этом они имеют различные химические свойства, а значит, и разное строение. Теория позволила доступно объяснить известные случаи изомерии. Бутлеров верил, что можно определить и пространственное расположение атомов в молекуле. Его предсказания были позже подтверждены, что дало толчок развитию нового раздела органической химии — стереохимии. Следует отметить, что ученый первым открыл и объяснил явление динамической изомерии. Ее смысл заключается в том, что два или несколько изомеров в определенных условиях могут легко переходить друг в друга. Если говорить в общем, то именно изомерия стала серьезным испытанием для теории химического строения и была ею блестяще объяснена.
Сформулированные Бутлеровым неопровержимые положения очень скоро принесли теории всеобщее признание. Верность выдвинутых идей была подтверждена экспериментами ученого и его последователей. В их процессе доказали гипотезу об изомерии: Бутлеров синтезировал один из четырех предсказанных теорией бутиловых спиртов, расшифровал его строение. В соответствии с правилами изомерии, которые напрямую вытекали из теории, также была высказана возможность существования четырех валериановых кислот. Позже они были получены.
Это лишь единичные факты в цепочке открытий: химическая теория строения органических соединений обладала потрясающей предсказательной способностью.
За относительно короткий период было открыто, синтезировано и изучено большое количество новых органических веществ и их изомеров. В итоге теория Бутлерова дала толчок бурному развитию химической науки, в том числе синтетической органической химии. Так, многочисленные синтезы Бутлерова являются главными продуктами целых отраслей промышленности.
Теория химического строения продолжила развиваться, что принесло органической химии много революционных по тем временам идей. К примеру, Кекуле выдвинул предположение о циклическом строении бензола и перемещении его двойных связей в молекуле, об особых свойствах соединений с сопряженными связями и многом другом. Более того, упомянутая теория сделала органическую химию более наглядной — появилась возможность рисовать формулы молекул.
А это, в свою очередь, положило начало классификации органических соединений. Именно использование структурных формул помогало определить пути синтеза новых веществ, установить строение сложных соединений, то есть обусловило активное развитие химической науки и ее отраслей. Например, Бутлеров стал проводить серьезные исследования процесса полимеризации. В России это начинание было продолжено его учениками, что в итоге позволило открыть промышленный способ получения синтетического каучука .
1.4: Развитие теории химической связи
Ключевые термины
Убедитесь, что вы можете определить и использовать в контексте следующие ключевые термины.
- прочность сцепления
- ковалентная связь
- ионная связь
- Структура Льюиса
- неподеленный электрон
- несвязывающий электрон
Учебные заметки
Чтобы успешно нарисовать структуры Льюиса, вам необходимо знать количество валентных электронов, присутствующих в каждом из задействованных атомов.Запомните количество валентных электронов, которыми обладает каждый из элементов, обычно встречающихся в органической химии: C, H, O, N, S, P и галогены.
Рисуя любую органическую структуру, вы должны помнить, что нейтральный атом углерода почти всегда будет иметь четыре связи. Точно так же водород всегда имеет одну связь; нейтральные атомы кислорода имеют две связи; а нейтральные атомы азота имеют три связи. Запомнив эти простые правила, вы сможете избежать ненужных ошибок в дальнейшем в ходе курса.
Тип представления «клин и ломаная линия», который помогает передать трехмерную природу органических соединений, будет использоваться на протяжении всего курса.
Обзор склеивания
Почему одни вещества представляют собой химически связанные молекулы, а другие — ассоциацию ионов? Ответ на этот вопрос зависит от электронной структуры атомов и природы химических сил внутри соединений. Хотя четко определенных границ нет, химические связи обычно подразделяются на три основных типа: ионные связи, ковалентные связи и металлические связи.В этой главе будет обсуждаться каждый тип связи и общие свойства типичных веществ, в которых встречается этот тип связи.
- Ионные связи являются результатом электростатических сил, которые существуют между ионами с противоположным зарядом . Эти связи обычно включают металл с неметаллом
- Ковалентные связи возникают в результате разделения электронов между двумя атомами . Связи обычно включают один неметаллический элемент с другим .
- Металлические связи обнаруживаются в твердых металлах (медь, железо, алюминий), причем каждый атом металла связан с несколькими соседними атомами металла, а связывающие электроны могут свободно перемещаться по трехмерной структуре.
Каждая классификация облигаций подробно обсуждается в следующих разделах главы. Давайте посмотрим на предпочтительное расположение электронов в атомах, когда они образуют химические соединения.
Рисунок 1.4.1 : Г. Н. Льюис и правило октета. (а) Льюис работает в лаборатории. (б) В первоначальном наброске правила октетов Льюис сначала поместил электроны по углам куба, а не поместил их, как мы делаем сейчас.
Символы Льюиса
В начале 20 -го века американский химик Г.Н. Льюис (1875–1946) разработал систему символов — теперь называемых символами электронных точек Льюиса, часто сокращенными до символов точек Льюиса — которые можно использовать для предсказания числа связей, образованных большинством элементов в их соединениях. Каждый символ точки Льюиса состоит из химического символа элемента, окруженного точками, которые представляют его валентные электроны.
Точек Льюиса:
- обеспечивают удобное представление валентных электронов
- позволяет отслеживать валентные электроны во время образования связи
- состоит из химического символа элемента и точки для каждого валентного электрона
Чтобы написать символ точки Льюиса элемента, мы размещаем точки, представляющие его валентные электроны, по одной вокруг химического символа элемента.Сверху, внизу, слева и справа от символа помещается до четырех точек (в любом порядке, при условии, что элементы с четырьмя или менее валентными электронами имеют не более одной точки в каждой позиции). Следующие точки для элементов с более чем четырьмя валентными электронами снова распределяются по одной, каждая в паре с одним из первых четырех. Фтор, например, с электронной конфигурацией [He] 2 s 2 2 p 5 , имеет семь валентных электронов, поэтому его точечный символ Льюиса строится следующим образом:
Рисунок \ (\ PageIndex {2} \): Точечные символы Льюиса для элементов периода 2
Льюис использовал непарные точки, чтобы предсказать количество связей, которые элемент будет образовывать в соединении.Рассмотрим символ азота на рисунке \ (\ PageIndex {2} \). Символ точки Льюиса объясняет, почему азот с тремя неспаренными валентными электронами имеет тенденцию образовывать соединения, в которых он разделяет неспаренные электроны с образованием трех связей. Бор, который также имеет три неспаренных валентных электрона в символе точки Льюиса, также имеет тенденцию образовывать соединения с тремя связями, тогда как углерод с четырьмя неспаренными валентными электронами в символе точки Льюиса имеет тенденцию делить все свои неспаренные валентные электроны, образуя соединения. в котором у него четыре облигации.Символы Льюиса — это инструмент, помогающий рисовать структуры. В следующем разделе мы увидим, почему связывание в молекулярных соединениях следует теории Льюиса.
Элементы того же периода имеют одинаковое количество валентных электронов и одинаковые символы Льюиса. Например, электронная конфигурация атомарной серы [Ne] 3s 2 3p 4 , таким образом, имеется шесть валентных электронов. Следовательно, его символ Льюиса будет похож на кислород и будет выглядеть так:
Правило октета
Главный вклад Льюиса в теорию связи заключался в признании того факта, что атомы имеют тенденцию терять, приобретать или делиться электронами, достигая в общей сложности восьми валентных электронов, называемых октетом .Это так называемое правило октетов объясняет стехиометрию большинства соединений в блоках s и p периодической таблицы. Теперь мы знаем из квантовой механики, что число восемь соответствует одной нс и трем валентным орбиталям np , которые вместе могут вместить в общей сложности восемь электронов. Примечательно, однако, что открытие Льюиса было сделано почти за десять лет до того, как Резерфорд предложил ядерную модель атома. Распространенными исключениями из правила октетов являются гелий, электронная конфигурация которого дает ему полную оболочку n = 1, и водород, который имеет тенденцию получать или делить свой один электрон для достижения электронной конфигурации гелия.
Идея Льюиса об октете объясняет, почему благородные газы редко образуют соединения. У них стабильная конфигурация s 2 p 6 (полный октет, бесплатно), поэтому у них нет причин реагировать и изменять свою конфигурацию. Все остальные элементы пытаются получить, потерять или разделить электроны для достижения конфигурации благородного газа. Это объясняет, почему атомы объединяются в соединения. Образуя связь, он делает атомы более стабильными и более низкими по энергии. Создание связей высвобождает энергию и представляет собой движущую силу для образования соединений.
Атомы часто приобретают, теряют или делят электроны, чтобы достичь того же количества электронов, что и ближайший к ним благородный газ в периодической таблице.
Льюис Структурс
Структуры Льюиса представляют, как символы Льюиса получают, теряют или делятся электронами для получения октета путем образования соединений.
Структуры Льюиса ионных соединений
Всякий раз, когда в структуре органического соединения присутствует металл, высока вероятность того, что присутствует по крайней мере одна ионная связь.Ионные связи представлены в структурах Льюиса иначе, чем ковалентные связи. Следует проявлять особую осторожность при рисовании структуры Льюиса органических соединений, содержащих ионную связь. Ионные связи обычно образуются, когда металл и неметалл являются частью соединения. Некоторые атомы достигают октета, полностью приобретая или теряя электроны для образования ионов. Ионные связи образуются за счет электростатического притяжения созданных ионов. Формула поваренной соли — NaCl. Это результат связывания ионов Na + и ионов Cl —.Если металлический натрий и газообразный хлор смешиваются в правильных условиях, они образуют соль. Натрий теряет электрон, а хлор получает этот электрон. При этом выделяется большое количество света и тепла. Полученная соль в основном нереактивна — она стабильна. Он не будет подвергаться взрывным реакциям, в отличие от натрия и хлора, из которых он сделан. Почему? Ссылаясь на правило октетов, атомы пытаются получить электронную конфигурацию благородного газа, которая состоит из восьми валентных электронов. Натрий (1s 2 2s 2 2p 6 3s 1 ) имеет один валентный электрон, поэтому отказ от него приведет к такой же электронной конфигурации, что и неон (1s 2 2s 2 2p 6 ).Хлор (1s 2 2s 2 2p 6 3s 2 3p 7 ) имеет семь валентных электронов, поэтому, если он берет один, у него будет восемь (октет). Хлор имеет электронную конфигурацию аргона (1s 2 2s 2 2p 6 3s 2 3p 8 ), когда он приобретает электрон.
Структура Льюиса ионного соединения показывает движение электронов. Для NaCl натрий находится в группе 1 и имеет один валентный электрон, а хлор находится в группе 17 и имеет семь валентных электронов.Натрий теряет свой единственный валентный электрон, тем самым становясь положительно заряженным. Хлор получает этот электрон, получая полный октет и отрицательный заряд. После усиления / потери электрона новые структуры Льюиса Na + и Cl — записываются рядом друг с другом, представляя ионную связь в NaCl.
Примеры структур Льюиса ионных соединений
Ковалентные связи и структуры Льюиса молекулярных соединений
В то время как щелочные металлы (такие как натрий и калий), щелочноземельные металлы (такие как магний и кальций) и галогены (такие как фтор и хлор) часто образуют ионы для достижения полного октета, основные элементы органической химии: углерод, водород, азот и кислород — вместо этого имеют тенденцию заполнять свой октет на , разделяя электронов с другими атомами, образуя ковалентные связи.Рассмотрим простейший случай газообразного водорода. Изолированный атом водорода имеет только один электрон, расположенный на орбитали 1 s . Если два атома водорода подходят достаточно близко, так что их соответствующие орбитали 1 s перекрываются, два электрона могут быть разделены между двумя ядрами, и образуется ковалентно связанная молекула H 2 . В структуре Льюиса H 2 каждая пара электронов, разделяемая между двумя атомами, изображена как одна линия, обозначающая одинарную ковалентную связь.
Водород представляет собой особый случай, потому что атом водорода не может соответствовать правилу октетов; ему нужно всего два электрона, чтобы иметь полную оболочку. Это часто называют «правилом дублета» для водорода.
Одной из простейших органических молекул является метан с молекулярной формулой CH 4 . Метан — это «природный газ», сжигаемый в домашних печах и водонагревателях, а также на электростанциях. Чтобы проиллюстрировать ковалентную связь в метане с использованием структуры Льюиса, мы сначала должны признать, что, хотя атом углерода имеет в общей сложности шесть электронов, символ Льюиса имеет четыре неспаренных электрона.Следуя теории Льюиса, атом углерода хочет образовать четыре ковалентные связи, чтобы заполнить свой октет. В молекуле метана центральный атом углерода делит свои четыре валентных электрона с четырьмя атомами водорода, таким образом образуя четыре связи и выполняя правило октетов (для углерода) и «правило дублета» (для каждого из атомов водорода).
Следующей относительно простой органической молекулой, которую следует рассмотреть, является этан, который имеет молекулярную формулу C 2 H 6 . Если мы нарисуем символ Льюиса каждого атома отдельно, мы увидим, что правило октета / дублета может быть выполнено для всех из них, образуя одну связь углерод-углерод и шесть связей углерод-водород.
Такой же подход можно использовать для молекул, в которых нет атома углерода. В молекуле воды символ Льюиса атома кислорода имеет два неспаренных электрона. Они связаны с одним электроном в символах Льюиса двух ковалентных связей O-H атомов водорода. Остальные четыре несвязывающих электрона на кислороде называются «неподеленными парами».
Поскольку электроны неподеленной пары часто НЕ отображаются в химических структурах, важно мысленно увидеть сложение неподеленных пар.Вначале может быть полезно физически добавить электроны неподеленной пары.
| метиламин | этанол | хлорметан |
Упражнение
Для следующей структуры введите все недостающие электроны неподеленной пары.
Ответ
Когда два или более электронов разделяются между атомами, образуется множественная ковалентная связь. Молекулярная формула этена (также известного как этилен, соединение, содержащееся в фруктах, таких как яблоки, сигнализирующее о созревании): C 2 H 4 . Расположив символы Льюиса для атомов, вы можете увидеть, что правило октета / дублета может выполняться для всех атомов, только если два атома углерода разделяют между собой двух пар электронов.Этен содержит двойную связь углерод / углерод.
Следуя этой схеме, тройная связь в молекулярной формуле этина C 2 H 2 (также известная как ацетилен, топливо, используемое в сварочных горелках) образуется, когда два атома углерода разделяют три пар электронов между их.
Упражнение \ (\ PageIndex {1} \)
Изобразите структуру Льюиса для аммиака, NH 3 .
- Ответ
Молекулярная форма
Чертеж метана палкой и клином показывает четырехгранные углы… (Клин выходит из бумаги, а пунктирная линия идет за листом. Сплошные линии находятся в плоскости бумаги.)
Следующие ниже примеры используют это обозначение, а также иллюстрируют важность включения несвязывающих электронных пар валентной оболочки при рассмотрении таких конфигураций.
| Метан | Аммиак | Вода |
Связующие конфигурации легко предсказываются теорией отталкивания электронных пар валентных оболочек, обычно называемой VSEPR в большинстве вводных текстов по химии.Эта простая модель основана на том факте, что электроны отталкиваются друг от друга, и разумно ожидать, что связи и несвязывающие пары валентных электронов, связанные с данным атомом, предпочтут находиться как можно дальше друг от друга. Конфигурации соединения углерода легко запомнить, так как их всего три категории.
| Конфигурация | Партнеры по связям | Соединительные уголки | Пример |
|---|---|---|---|
| Тетраэдр | 4 | 109.5º | |
| Тригональный плоский | 3 | 120º | |
| Линейный | 2 | 180º |
Рисунок 1.4.3
В трех показанных выше примерах центральный атом (углерод) не имеет несвязывающих валентных электронов; следовательно, конфигурация может быть оценена только по количеству партнеров по связыванию.Однако для молекул воды и аммиака несвязывающие электроны должны быть включены в расчет. В каждом случае есть четыре области электронной плотности, связанные с валентной оболочкой, так что ожидается тетраэдрический валентный угол. Измеренные валентные углы этих соединений (H 2 O 104,5º и NH 3 107,3º) показывают, что они ближе к тетраэдрическим, чем тригонально плоские или линейные. Конечно, именно конфигурация атомов (а не электронов) определяет форму молекулы, и в этом смысле аммиак называется пирамидальным (а не тетраэдрическим).Соединение трифторид бора, BF 3 , не имеет несвязывающих валентных электронов, а конфигурация его атомов является тригональной. Хорошие трактовки теории VSEPR были предоставлены Oxford и Purdue. Лучший способ изучить трехмерные формы молекул — использовать молекулярные модели. Студентам и профессиональным химикам доступно множество видов модельных наборов.
Две электронные группы
Наш первый пример — это молекула с двумя связанными атомами и без неподеленных пар электронов, Beh3.
AX
2 : BeH 21. Центральный атом, бериллий, дает два валентных электрона, а каждый атом водорода дает один. Электронная структура Льюиса —
Рисунок 1.4.4: Структура Льюиса для BeH 2
2. Вокруг центрального атома расположены две группы электронов. Из рисунка 1.4.3 видно, что расположение, минимизирующее отталкивание, размещает группы на 180 °.
3.Обе группы вокруг центрального атома представляют собой пары связей (BP). Таким образом, BeH 2 обозначается как AX 2 .
4. Из рисунка 1.4.3 мы видим, что с двумя парами связей молекулярная геометрия, которая минимизирует отталкивание в BeH 2 , составляет линейных .
AX
2 : CO 21. Центральный атом, углерод, дает четыре валентных электрона, а каждый атом кислорода дает шесть. Электронная структура Льюиса —
2.Атом углерода образует две двойные связи. Каждая двойная связь представляет собой группу, поэтому вокруг центрального атома есть две группы электронов. Как и в BeH 2 , конструкция, которая сводит к минимуму отталкивание, размещает группы на 180 °.
3. Еще раз, обе группы вокруг центрального атома представляют собой пары связей (BP), поэтому CO 2 обозначается как AX 2 .
4. VSEPR распознает только группы вокруг центрального атома . Таким образом, неподеленные пары на атомах кислорода не влияют на геометрию молекулы.С двумя связующими парами на центральном атоме и отсутствием неподеленных пар молекулярная геометрия CO 2 является линейной (рис. 1.4.3).
Три электронные группы
AX
3 : BCl 31. Центральный атом, бор, дает три валентных электрона, а каждый атом хлора дает семь валентных электронов. Электронная структура Льюиса —
2. Вокруг центрального атома расположены три группы электронов. Чтобы свести к минимуму отталкивание, группы располагаются на 120 ° друг от друга (рисунок 1.4.3).
3. Все электронные группы являются связующими парами (BP), поэтому структура обозначена как AX 3 .
4. Из рисунка 1.4.3 мы видим, что с тремя парами связей вокруг центрального атома молекулярная геометрия BCl 3 составляет тригонально плоских .
AX
3 : CO 3 2−1. Центральный атом углерода имеет четыре валентных электрона, а каждый атом кислорода имеет шесть валентных электронов. Как вы узнали ранее, электронная структура Льюиса одной из трех резонансных форм представлена как
2.Структура CO 3 2- представляет собой резонансный гибрид. Он имеет три идентичные связи, каждая с порядком связи 4/3. Мы минимизируем отталкивание, размещая три группы на расстоянии 120 ° друг от друга (рис. 1.4.3).
3. Все электронные группы являются связующими парами (БП). Структура с тремя связующими группами вокруг центрального атома обозначена как AX 3 .
4. Из рисунка 1.4.3 видно, что молекулярная геометрия CO 3 2- является тригональной плоской.
В нашем следующем примере мы впервые сталкиваемся с влиянием неподеленных пар и кратных связей на геометрию молекулы.
AX
2 E: SO 21. Центральный атом, сера, имеет 6 валентных электронов, как и каждый атом кислорода. Электронная структура Льюиса с 18 валентными электронами показана ниже.
2. Вокруг центрального атома расположены три группы электронов, две двойные связи и одна неподеленная пара.Сначала мы размещаем группы в тригональной плоскости, чтобы минимизировать отталкивание (рис. 1.4.3).
3. Есть две пары соединений и одна неподеленная пара, поэтому структура обозначается как AX 2 E. Это обозначение имеет в общей сложности три пары электронов, две X и одну E. Поскольку неподеленная пара не используется двумя ядер, он занимает больше места рядом с центральным атомом, чем связующая пара (рис. 1.4.4). Таким образом, связывающие пары и неподеленные пары электростатически отталкиваются друг от друга в порядке BP – BP
2 мы имеем одно взаимодействие BP – BP и два взаимодействия LP – BP. 4. Геометрия молекулы описывается только положениями ядер, , а не положениями неподеленных пар. Таким образом, с двумя ядрами и одной неподеленной парой форма изогнута или V-образная , что можно рассматривать как тригональное плоское расположение с отсутствующей вершиной (рисунки 1.4.2.1 и 1.4.3).
Рисунок 1.4.4: Разница в пространстве, занимаемом неподеленной парой электронов и связующей парой
Как и в случае с SO 2 , эта составная модель распределения электронов и отрицательного электростатического потенциала в аммиаке показывает, что неподеленная пара электронов занимает большую область пространства вокруг атома азота, чем связующая пара электронов, которая является общей с водородом. атом.
Подобно неподеленным парам электронов, кратные связи занимают больше места вокруг центрального атома, чем одинарная связь, что может привести к тому, что другие углы связи будут несколько меньше ожидаемых. Это связано с тем, что множественная связь имеет более высокую электронную плотность, чем одинарная связь, поэтому ее электроны занимают больше места, чем электроны одинарной связи. Например, в такой молекуле, как CH 2 O (AX 3 ), структура которой показана ниже, двойная связь отталкивает одинарные связи сильнее, чем одинарные связи.Это вызывает отклонение от идеальной геометрии (валентный угол H – C – H 116,5 °, а не 120 °).
Теории химической связи и ее истинная природа
Берсон, Дж. А .: Химическое творчество. Уайли, Вайнхайм (1999)
Google Scholar
Кисть, С .: Динамика изменения теории в химии. Stud. Hist. Филос. Sci. 30 , 263–302 (1999). DOI: 10.1016 / S0039-3681 (98) 00028-4
Артикул Google Scholar
Батлер, Л.: Роберт С. Малликен и научная политика. Stud. Hist. Филос. Sci. 25 , 25–45 (1994)
Google Scholar
Коулсон, К.А., МакВини, Р.: Валенсия Коулсона, 3-е изд. Oxford University Press, Оксфорд, Нью-Йорк (1979)
Google Scholar
Гавроглу К., Симойнс А .: Немцы, американцы и начало квантовой химии: слияние расходящихся традиций.Stud. Hist. Филос. Sci. 25 , 47–110 (1994)
Google Scholar
Hettema, H .: Quantum Chemistry. World Scientific, Сингапур (2000)
Google Scholar
Леннард-Джонс, Дж. Э .: Электронная структура некоторых двухатомных молекул. Пер. Faraday Soc. 25 , 668–686 (1929)
Артикул Google Scholar
Малликен, Р.С .: Присвоение квантовых чисел электронам в молекулах. Phys. Ред. 32 , 186–222 (1928)
Артикул Google Scholar
Малликен Р.С.: Жизнь ученого, стр. 213. Шпрингер, Берлин (1989)
Google Scholar
Парк, Б.С.: В «контексте педагогики»: изменение стратегии обучения и теории квантовой химии. В: Кайзер, Д. (ред.) Педагогика и практика науки, стр.287–319. MIT Press, Кембридж (2005)
Google Scholar
Полинг, Л .: Природа химической связи. Издательство Корнельского университета, Итака (1960)
Google Scholar
Полинг, Л., Уилсон, Э.Б .: Введение в квантовую механику. Dover Publications, Нью-Йорк (1963)
Google Scholar
Платт, Дж.Р .: Нобелевский лауреат по химии: Роберт С. Малликен. Наука 154 , 747 (1966)
Статья Google Scholar
Simões, A., Gavroglu, K .: Квантовая химия как прикладная математика. Вклад Чарльза Альфреда Коулсона (1910–1974). Hist. Stud. Phys. Биол. 29 , 363–406 (1999)
Google Scholar
Слейтер, Дж. К. Квантовая теория молекул и твердых тел, т.1. Макгроу-Хилл, Нью-Йорк (1963)
Google Scholar
Вемулапалли Г.К .: Редукция собственности в химии: некоторые уроки. Аня. Акад. Sci. 988 , 90–99 (2003)
Артикул Google Scholar
Вулли Р.Г .: Загадка молекулярной структуры. J. Chem. Educ. 62 , 1082 (1985)
Артикул Google Scholar
5.1 Теория валентной связи — Химия: сначала атомы 2e
Цели обучения
К концу этого раздела вы сможете:
- Опишите образование ковалентных связей в терминах перекрытия атомных орбиталей
- Определите и приведите примеры σ- и π-связей
Как мы знаем, научная теория — это убедительное объяснение наблюдаемых законов природы или больших массивов экспериментальных данных. Чтобы теория была принята, она должна объяснять экспериментальные данные и уметь предсказывать поведение.Например, теория VSEPR получила широкое распространение, поскольку она предсказывает трехмерные молекулярные формы, которые согласуются с экспериментальными данными, собранными для тысяч различных молекул. Однако теория VSEPR не дает объяснения химической связи.
Перекрытие атомных орбиталей
Существуют успешные теории, описывающие электронную структуру атомов. Мы можем использовать квантовую механику, чтобы предсказать конкретные области вокруг атома, где, вероятно, будут находиться электроны: сферическую форму для орбитали s , форму гантели для орбитали p и так далее.Однако эти предсказания описывают только орбитали вокруг свободных атомов. Когда атомы связываются, образуя молекулы, атомных орбиталей недостаточно для описания областей, в которых в молекуле будут располагаться электроны. Более полное понимание распределения электронов требует модели, которая может объяснить электронную структуру молекул. Одна популярная теория утверждает, что ковалентная связь образуется, когда пара электронов разделяется двумя атомами и одновременно притягивается ядрами обоих атомов.В следующих разделах мы обсудим, как такие связи описываются теорией валентных связей и гибридизацией.
Теория валентной связи описывает ковалентную связь как перекрытие наполовину заполненных атомных орбиталей (каждая из которых содержит один электрон), которые дают пару электронов, совместно используемых двумя связанными атомами. Мы говорим, что орбитали двух разных атомов перекрываются, когда часть одной орбитали и часть второй орбитали занимают одну и ту же область пространства. Согласно теории валентной связи, ковалентная связь возникает, когда выполняются два условия: (1) орбиталь на одном атоме перекрывает орбиталь на втором атоме и (2) отдельные электроны на каждой орбитали объединяются, образуя электронную пару.Взаимное притяжение между этой отрицательно заряженной электронной парой и положительно заряженными ядрами двух атомов служит для физического связывания двух атомов посредством силы, которую мы определяем как ковалентную связь. Сила ковалентной связи зависит от степени перекрытия вовлеченных орбиталей. Орбитали, которые сильно перекрываются, образуют связи, которые сильнее, чем те, которые перекрываются меньше.
Энергия системы зависит от того, насколько перекрываются орбитали. На рис. 5.2 показано, как сумма энергий двух атомов водорода (цветная кривая) изменяется по мере их приближения друг к другу.Когда атомы находятся далеко друг от друга, перекрытия нет, и по соглашению мы устанавливаем сумму энергий равной нулю. Когда атомы движутся вместе, их орбитали начинают перекрываться. Каждый электрон начинает ощущать притяжение ядра в другом атоме. Кроме того, электроны начинают отталкивать друг друга, как и ядра. Хотя атомы все еще далеко друг от друга, притяжение немного сильнее, чем отталкивание, и энергия системы уменьшается. (Начинает формироваться связь.) По мере того, как атомы сближаются, перекрытие увеличивается, поэтому притяжение ядер для электронов продолжает увеличиваться (как и отталкивание между электронами и между ядрами).На некотором определенном расстоянии между атомами, которое варьируется в зависимости от задействованных атомов, энергия достигает минимального (наиболее стабильного) значения. Это оптимальное расстояние между двумя связанными ядрами — это расстояние связи между двумя атомами. Связь стабильна, потому что в этот момент силы притяжения и отталкивания объединяются, чтобы создать минимально возможную энергетическую конфигурацию. Если бы расстояние между ядрами еще больше уменьшилось, отталкивание между ядрами и отталкивание, когда электроны удерживаются в более близкой близости друг к другу, стали бы сильнее сил притяжения.Затем энергия системы возрастет (что приведет к дестабилизации системы), как показано в крайнем левом углу рисунка 5.2.
Рис. 5.2 (a) Взаимодействие двух атомов водорода изменяется в зависимости от расстояния. (б) Энергия системы изменяется при взаимодействии атомов. Самая низкая (наиболее стабильная) энергия наблюдается на расстоянии 74 пм, что является длиной связи, наблюдаемой для молекулы H 2 .
Помимо расстояния между двумя орбиталями, ориентация орбиталей также влияет на их перекрытие (кроме двух орбиталей s , которые являются сферически симметричными).Большее перекрытие возможно, когда орбитали ориентированы так, что они перекрываются на прямой линии между двумя ядрами. Рисунок 5.3 иллюстрирует это для двух орбиталей p от разных атомов; перекрытие больше, когда орбитали перекрываются конец в конец, а не под углом.
Рис. 5.3 (a) Перекрытие двух орбиталей p является наибольшим, когда орбитали направлены встык. (b) Любая другая договоренность приводит к меньшему дублированию. Точки указывают расположение ядер.
Перекрытие двух орбиталей s (как в H 2 ), перекрытие орбитали s и орбитали p (как в HCl), а также сквозное перекрытие двух орбиталей Орбитали p (как в Cl 2 ) все создают сигма-связи (σ-связи), как показано на рисунке 5.4. Связь σ представляет собой ковалентную связь, в которой электронная плотность сосредоточена в области вдоль межъядерной оси; то есть линия между ядрами проходила бы через центр области перекрытия.Одинарные связи в структурах Льюиса описываются как σ-связи в теории валентных связей.
Рис. 5.4 Сигма (σ) связи образуются в результате перекрытия следующих элементов: (a) две орбитали s , (b) орбиталь s и орбиталь p и (c) две орбитали p . Точки указывают расположение ядер.
Пи-связь (π-связь) представляет собой тип ковалентной связи, которая возникает в результате параллельного перекрытия двух орбиталей p , как показано на рисунке 5.5. В π-связи области перекрытия орбиталей лежат по разные стороны от межъядерной оси. Вдоль самой оси расположен узел, то есть плоскость, на которой нет вероятности найти электрон.
Рис. 5.5 Связи Pi (π) образуются в результате параллельного перекрытия двух орбиталей p . Точки указывают расположение ядер.
В то время как все одинарные связи являются связями σ, кратные связи состоят как из связей σ, так и π. Как следует из приведенных ниже структур Льюиса, O 2 содержит двойную связь, а N 2 содержит тройную связь.Двойная связь состоит из одной σ-связи и одной π-связи, а тройная связь состоит из одной σ-связи и двух π-связей. Между любыми двумя атомами первая образованная связь всегда будет σ-связью, но в любом месте может быть только одна σ-связь. В любой множественной связи будет одна связь σ, а оставшиеся одна или две связи будут связями π. Эти связи описаны более подробно далее в этой главе.
Пример 5.1
Подсчет σ- и π-связей
Бутадиен, C 4 H 6 , используется для производства синтетического каучука.Определите количество σ- и π-связей, содержащихся в этой молекуле.
Решение
Имеется шесть связей σ C – H и одна связь σ C – C, всего семь из одинарных связей. Есть две двойные связи, каждая из которых имеет связь π в дополнение к связи σ. Это дает всего девять σ и две π связи.Проверьте свои знания
Обозначьте каждую иллюстрацию как изображающую связь σ или π:(a) параллельное перекрытие 4 p и 2 p орбитальных
(b) сквозное перекрытие 4 p и 4 p орбитальных
(c) сквозное перекрытие орбиты 4 p и 2 p орбиты
Ответ:
(a) — это π-связь с узлом вдоль оси, соединяющей ядра, а (b) и (c) — σ-связи, которые перекрываются вдоль оси.
Дипольные моменты и ионный характер
Теперь, когда мы увидели важность понимания связи между местоположением электронов в атомах и свойствах элементов, мы можем расширить наше понимание связь между атомами. Это будет введение в более сложные аспекты химическая связь, которая составляет основу самой химии. За исключением знатных В газах атомы сами по себе не обладают наиболее стабильной возможной электронной конфигурацией.Что Здесь само понятие химической связи вступает в силу: атомы могут достигать стабильной конфигурации путем обмена электронами с другим атомом, что приводит к образованию ионов.
Ионы, в свою очередь, могут связываться посредством заряда — простого кулоновского притяжения — что приводит к образованию соединений, которые мы называем ионными соединениями. Посмотрим на ионную природу связей. во-первых, с точки зрения простого положительно-отрицательного притяжения. Не менее важно то, что некоторые атомы связь путем обмена, а не обмена электронами; обмен электронами приводит к Ковалентная связь.Чтобы добавить еще одно измерение, некоторые химические вещества не являются полностью ионный или полностью ковалентный; эти виды обладают постоянным диполем и классифицируются как полярный.
В своем вводном курсе физики вы, вероятно, обсудите концепцию кулоновской взаимодействия в гораздо более строгих деталях, чем мы будем делать здесь. Нас интересуют в первую очередь различия в свойствах между видами, обусловленные их относительными ковалентными, ионными или полярными природа — не в строгой модели этих свойств.Мы озабочены подключением между потенциальной энергией и силой и относительным разделением (или отсутствием разделения) между обвинения. Начнем с определения электрической или кулоновской силы как произведения зарядов деленное на квадрат расстояния между этими зарядами:
Здесь Q принимается как фундаментальная постоянная заряда электрона: один электрон имеет заряд 1.60218 × 10−19C 1.60218 × 10−19C. (Мы будем работать исключительно в системе СИ, поэтому расстояния будут измеряться в метров (м)).
И, как вы помните, энергия — это сила, умноженная на расстояние, поэтому
Чтобы проиллюстрировать тенденцию силы притяжения, мы сначала рассмотрим силу притяжения между двумя ионы с одним зарядом, разделенные расстоянием 2 d :
F = (1) (- 1) (2d) 2 = −14d2F = (1) (- 1) (2d) 2 = −14d2И затем сила притяжения между двумя ионами с двойным зарядом, разделенными расстоянием d :
F = (2) (- 2) (d) 2 = −4d2F = (2) (- 2) (d) 2 = −4d2сила притяжения увеличивается с зарядом и уменьшается с увеличением расстояния.Если бы вся материя состояла из ионов, это был бы конец истории, но это явно не так. Там молекулы, у которых заряд — положительный или отрицательный — постоянно сконцентрирован больше на одном атоме, чем на другом. Обратите внимание, мы говорим атом, потому что эти соединения не состоит из ионов, а скорее из атомов, разделяющих электроны ковалентными связями.
Дипольные моменты Бонда
Концепция дипольного момента связи позволяет нам исследовать частичное разделение заряда между атомами.Это простая модель в применении к двухатомным молекулам, которая будет более чем достаточно для наших целей. Дипольный момент связи определяется как заряд, умноженный на расстояние — заряд снова измеряется кратным заряду электрона, или кулоны. Расстояние всегда будет в метрах. Поскольку мы рассматриваем очень маленькие зарядов и расстояний, и потому что это относительное разделение зарядов, а не фактическое значение для него, которое нас интересует, мы представим новый блок под названием Дебай, названный в честь физико-химик Питер Дебай:
1Дебай (D) = 3.336 × 10−30C-mμ = Q × d1Debye (D) = 3,336 × 10−30C-mμ = Q × d Полезность блока Debye будет показана на примере:
Для HCl дипольный момент связи, как известно, равен 1,08 D
Для HI дипольный момент связи, как известно, составляет 0,44 D
Сравнивая эти два, мы видим, что HI менее полярен, чем HCl, что и следовало ожидать. на основе значений электроотрицательности.
Мы сделали переход между концепцией ионного соединения и частично ионного соединения. один. Конечно, частично ионное соединение также по определению должно быть частично ковалентным.
Частично ионный характер
Концепция дипольного момента связи помогает объединить концепции ионной и ковалентной связи. Поскольку существует менее полное разделение зарядов, чем в ионной связи, мы можем называют полярные связи частично ионными по природе. В отличие от хлорида натрия водород хлорид показывает частичные заряды (обозначенные дельта-обозначением) на водороде и хлоре. В виде как и следовало ожидать от значений электроотрицательности, водород несет частичный положительный заряд, в то время как хлор несет частичный отрицательный заряд.Откуда берутся эти обвинения?
Частичные заряды легко вычислить, сравнив действительные дипольные моменты. (что может быть получено экспериментально с помощью спектроскопии) с ожидаемым диполем в предельный случай (т. е. если рассматривать молекулу ионной). Фактический дипольный момент равен 1.03 D.
Пример 5.2
Определение частичного ионного характера
Каковы частичные заряды молекулы HCl, длина связи которой составляет 0,127 нм?Решение
Дипольный момент связи равен (1.60218 × 10–19C) (0,127 × 10–9 м) (1,60218 × 10–19C) (0,127 × 10–9 м) или 2,03 × 10–29C-м2. 03 × 10–29C-м. Преобразовано в D, это (2,03 × 10-29C-м) (1Дебай3,336 × 10-30C-м) (2,03 × 10-29C-м) (1Дебай3,336 × 10-30C-м) или 6,09 D. Была ли HCl полностью ионной. , это был бы его молекулярный диполь момент. Чтобы получить частичный ионный характер, разделим экспериментально измеренный момент связи на это предельное значение: % ионный характер = µexpµlim × 100% = (1.03D (6.09D) × 100% = 17 %% ionic характер = µexpµlim × 100% = (1.03D (6.09D) × 100% = 17%. Это означает, что связь составляет около 17% ионной — или, другими словами, положительный заряд в H равен +0.17 и частичный отрицательный заряд по хлору, –0,17.Проверьте свои знания
Повторите расчет для HI, который имеет дипольный момент 0,42 D и длину связи 0,161 нм.Ответ: Вычислено 7,73, процентов 5,43
Что можно сказать об относительной полярности связи HI по сравнению с полярностью связи HCl? Согласуется ли рассчитанный дипольный и процентный ионный характер с разницей в электроотрицательность между Cl и I?
Электронная конфигурация атома или иона является ключом к пониманию химического поведение элемента.Атомы, составляющие элемент, комбинируются по-разному, в диапазоне от преимущественно ионного (NaCl) до частично ионного (HCl) до того, что мы будем называть чисто ковалентным. На самом фундаментальном уровне все химические связи включают электроны, и значительный процент химических и физических свойств можно объяснить, учитывая расположение и разделение заряда в виде. Поняв структуру материи на атомном уровне, мы можем начать чтобы понять поведение материи как на микроскопическом, так и на макроскопическом уровне. уровни.
Понимание диполей и частичного ионного характера является фундаментальным для понимания взаимодействия между частицами, которые мы рассмотрим в главе о жидкостях и твердых телах. Эти межмолекулярные силы становятся важными в жидком и твердом состояниях вещества.
Этот сайт использует файлы cookie для повышения производительности. Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка вашего браузера для приема файлов cookie
Существует множество причин, по которым cookie не может быть установлен правильно.Ниже приведены наиболее частые причины:
- В вашем браузере отключены файлы cookie. Вам необходимо сбросить настройки браузера, чтобы он принимал файлы cookie, или чтобы спросить вас, хотите ли вы принимать файлы cookie.
- Ваш браузер спрашивает вас, хотите ли вы принимать файлы cookie, и вы отказались. Чтобы принять файлы cookie с этого сайта, нажмите кнопку «Назад» и примите файлы cookie.
- Ваш браузер не поддерживает файлы cookie. Если вы подозреваете это, попробуйте другой браузер.
- Дата на вашем компьютере в прошлом.Если часы вашего компьютера показывают дату до 1 января 1970 г., браузер автоматически забудет файл cookie. Чтобы исправить это, установите правильное время и дату на своем компьютере.
- Вы установили приложение, которое отслеживает или блокирует установку файлов cookie. Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.
Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу.Чтобы предоставить доступ без файлов cookie потребует, чтобы сайт создавал новый сеанс для каждой посещаемой страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файле cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в cookie; никакая другая информация не фиксируется.
Как правило, в файлах cookie может храниться только информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта.Например, сайт не может определить ваше имя электронной почты, пока вы не введете его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступа к остальной части вашего компьютера, и только сайт, который создал файл cookie, может его прочитать.
Теория информации, атомы в молекулах и молекулярное подобие
Реферат
Используя теорию информации, утверждается, что среди возможных определений того, что такое атом, когда он находится в молекуле, особое внимание заслуживает конкретное.А именно, это атом, определяемый «разделением акционеров» молекулы, изобретенной Хиршфельдом [(1977) Theor. Чим. Acta 44, 129]. Используемый теоретический инструмент — принцип минимального дефицита энтропии (принцип минимума недостающей информации) Кульбака и Либлера [(1951) Ann. Математика. Стат. 22, 79]. Соответствующий анализ дается проблеме оценки сходства между молекулами или частями молекул.
В основе химии лежит понимание молекул как комбинаций атомов.Поэтому неудивительно, что концепция атомов в молекулах (AIM) много обсуждалась в литературе (1). Химия в основном включает небольшие изменения между атомами и молекулярными фрагментами с достаточно хорошо понятными молекулярными инвариантами, например AIM, функциональные группы, молекулярные подсистемы и т. Д., Которые стремятся сохранить свою идентичность. Большинство молекулярных систем можно представить как состоящие из слегка возмущенных атомов (или атомных ионов), возможно, деформированных присутствием молекулярных остатков и демонстрирующих модифицированные чистые заряды, возникающие в результате переноса заряда и / или образования химических связей.Следовательно, эти химические атомы являются открытыми подсистемами.
Впечатляющее разнообразие опубликованных теоретических методов разделения молекулярной плотности на вклады AIM, например ссылки 1–15, свидетельствует о важности этой темы для химии. Различные методы основаны на разных принципах, некоторые в определенной степени произвольны (5) или эвристические (15), которые могут привести к противоречивым тенденциям в связанных атомных чистых зарядах (эффективных степенях окисления). Методы различаются используемой теоретической техникой, например.g., топологический анализ плотности, описание волновой функции или описание функционала плотности. Они также различаются используемыми физическими / эвристическими принципами, например, выравниванием электроотрицательности, нулевым потоком и правилами минимальной энергии продвижения. У них могут быть определенные недостатки, например, зависимость от базисного набора. Хорошо известный и привлекательный квантово-топологический подход (1–4) страдает от того факта, что определенные им атомные плотности не являются «υ-представимыми». [Атомная плотность является υ-представимой, когда существует внешний потенциал, который имеет эту плотность как плотность основного состояния.]
Соответствующие плотности изолированных атомов являются хорошими справочными материалами для определения свойств химических атомов в терминах быстро сходящихся рядов Тейлора по внешнему потенциалу и смещениям переноса заряда относительно потенциалов и зарядов свободных атомов. Сумма изолированных плотностей атома / иона с ядерными каспами в фактических положениях ядер определяет распределение плотности промолекулы (15). Промолекула является ключевым ингредиентом в анализе разности плотностей химической связи, проведенном Хиршфельдом (15), в котором предполагается, что при формировании молекулы каждый атом участвует в локальном усилении или потере пропорционально его локальному вкладу в плотность промолекулы.В данной статье мы восстановим это «разделение запаса» по Хиршфельду молекулярной плотности на атомарные компоненты.
Можно было бы надеяться обнаружить, что химический атом, как и его свободный аналог, будет обладать единственным острием в электронной плотности, связанным с эффективным атомным номером ядра (16). Плотности AIM должны быть связаны как с плотностями промолекул, так и с молекулярными плотностями, как представляющие атомные фрагменты в конкретной молекулярной системе. Хотелось бы некоторой степени перекрытия между плотностями этих химических атомов, чтобы отразить наличие химических связей (1, 17, 18).
То, что основные состояния атомов и / или их небольшие возмущения являются однозначно подходящими эталонными состояниями для подробного описания AIM, было хорошо понято пионерами, например Полингом и Малликеном, при использовании ими концепций гибридизации, продвижения и т. Д. поляризация и ионный характер. В идеале определение AIM сохранит как можно больше информации о разделенных атомах. [Здесь и далее слово «атомы» обычно означает атомы или ионы.]
При определении AIM, как можно сохранить, насколько это возможно, информационное содержание атомов в основном состоянии? Для этой цели естественно использовать некоторый информационно-теоретический принцип (19–26).И здесь помогает теория функционала плотности (ДПФ) (27, 28). Для DFT утверждает, что сама электронная плотность несет всю информацию об основном состоянии. Итак, мы можем определить AIM таким образом, чтобы атомные плотности были максимально похожи на плотности изолированных атомов, и, таким образом, получить «лучшие» атомы, которые мы можем иметь в молекуле в теоретико-информационном смысле.
Функционал Кульбака – Лейблера энтропийного дефицита
Предположим, что P 0 (r⃗) — заданное (эталонное) правильное распределение вероятностей, а P (r⃗) — некоторое пробное распределение вероятностей, информационное содержание которого мы хотим сделать как можно ближе к таковому из P 0 , с учетом одного или нескольких ограничений.Определите (25) функционал энтропийного дефицита (недостающей информации) как 1 и пусть существуют ограничения вида 2. Тогда P , которое является «наилучшим» приближением к P 0 , которое удовлетворяет ограничениям, получается путем решения принцип минимального дефицита энтропии (недостающая информация) 3, где {λ k } — множители Лагранжа. Δ S и λ k находятся путем решения уравнений 2 и 3 одновременно. Данный множитель Лагранжа измеряет, насколько чувствителен дефицит энтропии к соответствующему ограничению.Если существует несколько решений для Δ S , выбирается минимальное.
Эта ограниченная экстремизация энтропии (24) представляет собой наиболее беспристрастный способ усвоения (как можно более полного) информации, включенной в эталонное распределение и вспомогательные условия. Для конкретных P и P 0 , уравнение. 1 дает неотрицательное число, которое определяет информационное расстояние между двумя распределениями или дефицит энтропии P относительно P 0 .
Пример: Чтобы приступить к решению проблемы AIM, предположим, что известна точная плотность электронов AB, ρ, и что мы хотим представить ее как сумму двух плотностей ρ A и ρ B с номерами электронов N A 0 и N B 0 , оба фиксированные, и мы хотим сохранить ρ A как можно ближе к ρ A 0 и ρ B как можно ближе к ρ B 0 насколько возможно.Возьмем 4 и ограничения 5 с множителями Лагранжа λ A и λ B соответственно. Уравнения. 2 и 3 тогда дают 6 То есть, если «атомы» A и B замкнуты (им не разрешено переносить электроны), энтропия не приводит к изменению плотности. Чтобы определить атом в молекуле, нужно будет предполагать перенос электрона.
Деривация раздела акционеров Хиршфельда на AIM
Элементарная модификация только что приведенного примера дает то, что мы хотим, открытые атомы A и B с электронными номерами N A и N B , не обязательно равными N A 0 и N B 0 , но все еще соответствует N A + N B = N A 0 + N B 0 = N 0 .Мы можем потребовать исчерпывающего распределения всего электронного распределения в AB либо A, либо B, 7. Это ограничение требует использования множителя Лагранжа λ (r⃗), который является функцией r⃗. Здесь ρ обозначает точную (предполагаемую известную) плотность системы AB, которая нормирована на N 0 электронов. Нормирована также на N 0 изоэлектронная плотность промолекулы ρ 0 = ρ A 0 + ρ B 0 . Реализация принципа минимального дефицита энтропии уравнения.3, с ограничением уравнения. 7 дает уравнение для плотностей AIM ρ A и ρ B , 8, где 1 n D (r⃗) = λ (r⃗) -1. Следовательно, 9 Коэффициент пропорциональности D (r⃗) определяется из ограничения уравнения. 7: 10 Таким образом, мы находим 11. Это «разделение запаса» электронной плотности, предложенное давно Хиршфельдом (7).
Фактор разделения w (r⃗) определяет относительную долю атома в плотности промолекулы ρ 0 (r⃗).Результат тривиально обобщается на любое количество составляющих атомов. «Атомы» могут быть нейтральными атомами, ионами или функциональными группами.
Минимизация энтропийного дефицита Кульбака – Либлера уравнения 8, как видно, обеспечивает прочную теоретическую основу для предписания Хиршфельда, которое, как известно, дает достаточно переносимые распределения зарядов и моменты (15,30), которые можно использовать, например, при расчетах электростатического потенциала и энергии взаимодействия. Фрагменты AIM, определенные формулой.11 имеют непрерывную, но хорошо локализованную плотность. Эта процедура разделения не зависит от базового набора и может использоваться как для теоретических, так и для экспериментальных плотностей.
Плотность AIM уравнения. 11 обладают правильным поведением на границе разделенных (изолированных) атомов, где все межъядерные расстояния R ∝β → ∞, потому что D (r⃗) → 1, когда ρ (r⃗) → ρ 0 (r⃗). Плотности AIM достаточно хорошо удовлетворяют, хотя и не идеально, надлежащим условиям ядерного каспа и дальнего распада.Эти плотности, скорее всего, являются υ-представимыми или, по крайней мере, ансамблевыми υ-представимыми. [Недавняя неопубликованная работа Пола Айерса по проблеме υ-представимости подразумевает, что атомные плотности Хиршфельда либо υ-представимы, либо бесконечно близки к υ-представимости.]
Фактор связи D (r) уравнения. 10 показывает, как изменилась плотность свободного атома в химическом атоме. В обычном случае, когда происходит накопление плотности в области молекулярных связей и истощение в несвязывающих областях атомов, связанных в молекуле, относительно ρ 0 , связанные атомы поляризованы по направлению к своим связывающим партнерам.
Каждый химический потенциал связанных атомов должен быть равен молекулярному химическому потенциалу μ (8, 9, 31). Аргумент следующий. Для фиксированного внешнего молекулярного потенциала υ имеем: 12, так что 13 14 15 Вычитая уравнение. 15 из уравнения. 14 дает: 16 Следовательно, если мы определим 17, получим: 18
AIM из модифицированного функционала энтропии
Чтобы проиллюстрировать множество возможностей для расширений и вариаций этого анализа, мы исследуем другой функционал энтропийного дефицита, в который, кроме того, мы включаем информационное расстояние между ρ (r̄) и результирующей молекулярной плотностью ρ̄ A (r⃗) + ρ̄ B (r⃗) ≡ ρ̄ (r⃗), который не является фиксированным, но подлежит определению.Модифицированный уравнение. 4 становится 19. Мы минимизируем это с учетом ограничения на фиксированное общее количество электронов, 20, где λ — глобальный множитель Лагранжа. Устанавливая 1 nc = λ −2, мы получаем оптимальную плотность AIM 21 и оптимальную общую плотность 22. Константа пропорциональности может быть определена из глобального ограничения в уравнении. 20: 23 Таким образом, вариационный принцип уравнения. 20 дает оптимальную молекулярную плотность ρ̄ (r⃗) как среднее геометрическое значение промолекулы и истинную плотность основного состояния молекулы.
Сравнение уравнения. 21 с уравнениями. 9 и 10 показывает, что эта модифицированная минимизация дефицита энтропии также дает химические атомы, которые должным образом поляризованы по направлению к связывающей области ковалентной связи. Однако этот эффект слабее в формуле. 21, потому что D̄ (r⃗) = [ D (r⃗)] 1/2 , где D (r⃗) (уравнение 10) обозначает фактор связи между атомами-держателями акций Хиршфельда. Тем не менее, обнадеживает то, что последний член «подобия» в формуле. 19, который движет ρ к ρ, как видно, изменяет плотность промолекулы ρ 0 в качественно правильном направлении, в сторону ρ, тем самым создавая частичную связь в молекуле.Плотность ρ̄ — это переходная плотность между ρ 0 и ρ.
Оба атома Хиршфельда уравнения. 11 и модифицированные химические атомы уравнения. 21 представляют собой открытые атомные фрагменты в молекуле, с эффективными зарядами 24, обычно отличными от заряда свободного атома (иона), q ∝ 0 = Z ∝ — N [ρ ∝ 0 ], вызванный компонентом переноса заряда химической связи. Однако в этом случае обычно не достигаются равные химические потенциалы.
Теоретико-информационная мера молекулярного сходства
Концепция молекулярного сходства иногда используется для характеристики структурного сходства различных молекул или их фрагментов. Используемые меры включают интегралы перекрытия для электронной плотности, электростатического потенциала и функции Фукуи (32). Здесь мы предполагаем, что подобие электронной структуры подразумевает близость информативности распределений электронной плотности.Это снова требует меры информационного расстояния, обеспечиваемого функционалом энтропийного дефицита уравнения. 1.
При типичном скрининговом поиске среди видов-кандидатов с плотностями {ρ i }, которые тестируются на химическую активность, аналогичную эталонной системе с плотностью ρ 0 , можно выбрать виды с минимальным значением энтропийного дефицита Δ S [ρ i | ρ 0 ]. Когда требуется реакционная способность определенного «активного центра», в качестве носителей входной информации следует выбирать плотности соответствующих молекулярных фрагментов.
Простейший вариационный критерий минимального дефицита энтропии между исследуемой плотностью молекулы / фрагмента ρ i и эталонной плотностью ρ 0 , нормированной на N [ρ 0 ] = N 0 , при условии ограничение нормализации N [ρ i ] = N i 0 , будет 25, что дает 26 Таким образом, наиболее благоприятное согласование достигается, когда функции формы P i (r⃗) и P 0 (r⃗) как можно ближе.Этот результат напоминает недавнее расширение теорем Хоэнберга – Кона (26) Эйерсом (33), демонстрирующее, что функция формы связанных состояний молекулы определяет все свойства рассматриваемой системы. Требование максимального количества общей информации в ρ i и ρ 0 действительно требует одинаковых функций формы для двух сравниваемых плотностей.
В качестве последнего примера рассмотрим критерий оптимального согласования пробной электронной плотности ρ с двумя эталонными плотностями ρ 1 0 ( N [ρ 1 0 ] = N 1 0 ) и ρ 2 0 ( N [ρ 2 0 ] = N 2 0 ), с учетом обычного ограничения нормализации, N [ρ] = N 0 : 27 Решение этого уравнения дает 28 Оптимальное соответствие — это нормализованное среднее геометрическое.
Такая проблема возникает при гетерогенном катализе, когда плотность адсорбата (ρ) согласовывается с двумя поверхностно-активными центрами (ρ 1 0 , ρ 2 0 ), участвующими в связывании адсорбата. Другой пример — связывание молекулы с двумя участками другой молекулы. Максимальное значение ρ (r⃗) ожидается в области перекрытия между ρ 1 0 и ρ 2 0 , потому что критерий наименьшего смещения информационного расстояния по формуле.27 требует, чтобы ρ напоминало (заметно перекрывалось) плотность перекрытия между двумя эталонными плотностями.
Резюме и заключительные замечания
В этой статье мы использовали теорию информации, чтобы разложить молекулярную электронную плотность на составляющие ее атомные плотности. В частности, мы выбрали подход Кульбака и Лейблера (25), основанный на дефиците энтропии (информационное расстояние), с использованием плотности свободных атомов в качестве эталонных распределений для получения плотностей AIM.Такая ссылка является одновременно естественной и уникальной в химии, потому что согласно теореме Хоэнберга-Кона изолированные плотности атомов несут всю информацию периодической таблицы, которая является источником химической мысли.
Мы продемонстрировали, что принцип минимального энтропийного дефицита (отсутствующей информации) при условии исчерпывающего разделения молекулярной плотности восстанавливает определение связанных атомов как «держателя запасов», сделанное Хиршфельдом (15), тем самым обеспечивая его фундаментальный теоретический вывод. .Эти химические атомы Хиршфельда представляют собой несмещенные части молекулярной плотности, которые наименее далеки по своему информационному содержанию от своих изолированных аналоговых атомов. Химические атомы выровняли химические потенциалы на глобальном, молекулярном уровне, и они, вероятно, являются υ-представимыми. Их перекрытие в молекуле согласуется с известной классической интерпретацией происхождения химической связи (17). Квантово-топологические атомы, напротив, не имеют перекрытия (1).
В расширении мы продемонстрировали, как функционал энтропийного дефицита может быть использован для создания связывающего характера химических атомов.Результирующая оптимальная плотность молекулы тогда определяется как среднее геометрическое истинной плотности основного состояния и плотности промолекулы (с плотностями свободных атомов, центрированными в фактических положениях ядер в молекуле). Обсуждаются как промежуточная поляризация (промотированная), так и конечная стадия переноса заряда реорганизации атомной плотности в молекуле. Мы также проиллюстрировали использование принципа минимизации энтропийного дефицита в задачах молекулярного подобия.
Наконец, отметим, что те же идеи применимы к возбужденным электронным состояниям.В этом случае, однако, эталонные плотности основных состояний свободных атомов могут не соответствовать минимальному значению информативного расстояния. Использование плотностей возбужденных состояний разделенных атомов в качестве эталонных плотностей может дать более реалистичные плотности возбужденных состояний (продвинутых) AIM, при этом все еще связывая полученные химические атомы с информацией об изолированных атомах (периодическая таблица). Теоретико-информационный подход должен быть полезен при количественных исследованиях переноса заряда и эффективных степеней окисления.
Благодарности
У нас были полезные обсуждения с несколькими людьми, в первую очередь с Полом Айерсом, Ричардом Бадером, Питером Политцером и Веден Смит. Это исследование было поддержано грантом Комитета научных исследований Польши и грантом Фонда нефтяных исследований Американского химического общества.
Аббревиатура
- AIM,
- атомов в молекулах
- Авторское право © 2000, Национальная академия наук
Структурная теория — обзор
2 Что такое механизм в органической химии?
Химики-органики используют термин «механизм» в двух разных смыслах, которые играют разные роли в том, как эти химики понимают химические превращения.Механизм в широком смысле — это, грубо говоря, полная характеристика динамического процесса преобразования набора молекул реагентов в набор молекул продуктов. 3 Такая характеристика отслеживает, как непрерывный путь, движения атомных ядер и — в некоторой форме — регистрирует зависящую от времени эволюцию электронов, которая совпадает с ядерными движениями. Следовательно, механизмы в этом широком смысле — это что-то вроде кинофильмов химического превращения. С другой стороны, механизмы в тонком смысле представляют собой дискретные характеристики преобразования как последовательности шагов.Начиная с реагентов (или, по крайней мере, некоторых из них), эти стадии описывают превращение одной важной структуры 4 по пути превращения в следующую важную структуру по пути. Иногда, отслеживая движение электронов стрелками, также предоставляется указание на то, как достигается преобразование между этими важными промежуточными соединениями. Таким образом, механизмы в этом тонком смысле похожи на серию тщательно отобранных и последовательно упорядоченных снимков химического превращения.Эти разные способы представления химического превращения в виде непрерывного процесса или дискретной последовательности шагов вписываются в теоретический аппарат органической химии в разных местах. Толстая концепция механизма реакции лежит в основе теоретических моделей химического превращения, взятых из термодинамики (таких как теория равновесия и спонтанности и теория переходного состояния). Тонкая концепция механизма связывает преобразование с классической структурной теорией органической химии, которая пытается объяснить характеристики химических превращений с точки зрения особенностей структур (или структурных формул) реагирующих частиц.Поскольку понятие механизма имеет эти два разных смысла, но при этом рассматривается как изображение (в любом смысле) одного и того же процесса, оно служит концептуальным мостом, связывающим классическую структурную теорию органической химии с более абстрактными моделями преобразований. .
Когда авторы учебников по органической химии от среднего до продвинутого дают характеристику понятия механизма, они часто начинают с некоторой версии толстой концепции. Гулд в своей классической книге «Механизм и структура в органической химии» описывает механизм реакции как: «гипотетическую картину поведения участвующих атомов.Такая картина, по-видимому, должна начаться за некоторое время до того, как реагирующие частицы приблизятся друг к другу, а затем продолжится для записи непрерывного пути атомов (и их электронов) во время реакции »[Gould, 1959, p. 127]. Миллер предлагает расплывчатую, но похожую характеристику: «Механизм химической реакции состоит из всего, что происходит, когда исходные материалы реакции превращаются в продукты» [Miller, 2004, p. 1]. Обе эти характеристики предполагают, что механизмы представляют собой или являются отображением непрерывного процесса преобразования между реагентами и продуктами.После введения этого толстого понятия механизма эти авторы сразу же отступают, заявляя, что химики, как правило, не способны предоставить и не нуждаются в таких полных и непрерывных характеристиках механизма. Вместо этого, самое большее, что химики могут установить или к чему они должны апеллировать, — это тонкое, дискретное понятие механизма. 5 Затем учебники переходят к описанию механизмов множества различных химических реакций, причем все в тонком смысле.Учитывая, что именно механизмы в тонком смысле слова занимают центральное место в изложении теории органической химии, стоит задуматься над тем, почему толстая концепция механизма продолжает играть важную роль в первоначальных характеристиках механизмов реакции химиками.
Один из очевидных способов объяснить непреходящую важность толстой концепции механизма состоит в том, что она представляет собой регулирующий идеал, к которому стремятся химики, но которого они не могут достичь.Если бы это объяснение было правильным — а это, по общему признанию, то, что часто предлагается в учебниках, — можно было бы ожидать, что можно будет идентифицировать то, что дополнительная информация, теоретически содержащаяся в подробном описании механизма, позволила бы химику-органику объяснить или предсказать дизайн, который он или она не может сделать с помощью простой тонкой характеристики механизма. Такое прагматическое следствие возможности дать подробное описание механизма придало бы некоторый реальный вес идее о том, что толстые описания — это то, к чему должен стремиться химик-органик.Хотя есть некоторые правдоподобные примеры, когда подробное описание механизма может оказаться важным для понимания (в смысле химиков-органиков) результата химической реакции, 6 обычно не так. Только в исключительных обстоятельствах для химика-органика будет иметь какой-либо практический смысл продолжить исследование механизма реакции после того, как он разработал убедительные аргументы в пользу тонкого описания механизма реакции.Это говорит о том, что, по крайней мере в практическом смысле, причина того, что химики-органики продолжают характеризовать механизмы в широком смысле, не в том, что они каким-то образом являются конечной целью механистических исследований. Вместо этого я думаю, что толстая концепция механизма играет более абстрактную, концептуальную роль, потому что она естественным образом вписывается в теоретические модели, которые химики используют для представления химических превращений.
Самый фундаментальный способ, которым химики-органики представляют химические превращения, — это пути вдоль поверхностей потенциальной энергии.В своей самой богатой версии поверхность потенциальной энергии отображает потенциальную энергию (или иногда свободную энергию) системы взаимодействующих молекул в зависимости от изменений относительного положения составляющих их атомов. Путь на этой поверхности от расположения атомов, соответствующих разделенным реагентам, к расположению атомов, соответствующих разделенным продуктам, будет записывать энергию химической системы, которая претерпевает непрерывное превращение из реагентов в продукты. Изменения положения атомов (и распределения электронов) вдоль пути с наименьшей энергией такого рода примерно соответствовали бы механизму реакции в широком смысле.Механизмы в этом широком смысле, таким образом, переводятся в математическое представление химического превращения в рамках фундаментальных рамок, которые используют химики-органики для размышлений о химических превращениях. Когда химические реакции рассматриваются как пути вдоль поверхности потенциальной энергии, появляется возможность использовать термодинамику и статистическую механику для разработки моделей, которые связывают скорости и распределения продуктов химического превращения, которые являются основными экспериментальными фактами, доступными химикам-органикам, с относительные разности энергии, представленные на поверхности потенциальной энергии.Таким образом, рассуждая о механизмах химических реакций в этом широком смысле, химики-органики могут установить взаимосвязь между экспериментальными фактами, которые они измеряют в лаборатории (скорости и распределения продуктов), и абстрактными математическими моделями химических превращений. Наблюдаемые особенности реакций накладывают ограничения на то, какой должна быть поверхность потенциальной энергии, соответствующая превращению, и наоборот. И объяснение, и предсказание результатов химических реакций, и экспериментальное определение механизмов зависят, таким образом, от использования этих взаимных ограничений.
Учитывая, что фундаментальные модели химических реакций рассматривают их как непрерывные превращения, тесно связанные с механизмами в широком смысле слова, может возникнуть недоумение, как химикам-органикам удается обходиться почти исключительно тонкими характеристиками механизмов реакций. В следующем разделе я представлю пример того, как такой механизм развивается и развивается перед лицом дополнительных экспериментальных данных, но можно сказать что-то о том, как это делается заранее.Основная идея состоит в том, что для объяснений и предсказаний, которыми в первую очередь занимаются химики-органики, необходимо знать лишь кое-что об относительных энергиях нескольких выбранных структур на непрерывном пути превращения реагентов в продукты. Пока описание механизма позволяет сделать вывод об этих важных структурах (обычно о переходном состоянии и стабильных промежуточных звеньях), оно будет поддерживать виды выводов, необходимых для предсказания и объяснения.Точно так же виды экспериментальных результатов, к которым обычно обращаются химики для установления механизма реакции, позволяют только сделать какие-то выводы об этих важных моментах на непрерывном пути превращения. Поэтому именно потому, что виды объяснений и предсказаний, произведенные химиками-органиками, требуют только энергетической информации о нескольких избранных структурах на пути трансформации, тонкие характеристики способны играть центральную роль в теории органической химии (см. [ Goodwin, 2003; 2007] для более подробных примеров того, как механизмы используются в такого рода объяснениях).
Учебники по органической химии обычно подчеркивают две вещи при описании (тонких) механизмов, которые фактически будут занимать их в основной части их текстов. Во-первых, механизмы реакции указывают или позволяют сделать вывод о структурах важных промежуточных структур на пути от реагентов к продуктам. Гулд, например, характеризует механизм в тонком смысле как «изображение участвующих видов в один или несколько критических моментов в ходе реакции» [Gould, 1959, p.127]. Это то, чего можно было ожидать, учитывая приведенное выше объяснение того, почему тонких механизмов достаточно для понимания органической химии. Во-вторых, эти механизмы разбивают химическую реакцию на последовательность этапов. Например, Лоури и Ричардсон говорят: «Механизм реакции — это спецификация посредством последовательности элементарных химических стадий подробного процесса, посредством которого происходит химическое изменение» [Lowry and Richardson, 1987, p. 190]. Эти две особенности механизмов связаны, и обе они важны для роли механизмов в понимании химических реакций.
Предоставление информации о важных промежуточных структурах в превращении, таких как переходное состояние или стабильные промежуточные соединения, позволяет химикам-органикам выполнять структурный анализ относительных энергий этих промежуточных продуктов. Структурный анализ, в свою очередь, позволяет предсказывать или объяснять относительные энергетические различия между структурами на основе небольшого набора надежно применимых и легко распознаваемых структурных особенностей. Эти относительные разности энергий затем можно использовать с помощью стандартных теоретических моделей химических переходов для вывода важных эмпирически измеримых характеристик химических реакций, таких как относительные скорости или распределения продуктов. 7 Таким образом, структурный анализ относительной разницы энергий между важными промежуточными соединениями в ходе химического превращения является современной реализацией цели Бутлерова объяснить все химические свойства веществ на основе их химической структуры. 8 Именно эта особенность механизмов облегчает применение классической структурной теории и ее современных потомков к современному пониманию химических реакций.
Разделение реакции на шаги имеет несколько важных функций.Он разбивает то, что может быть сложным структурным изменением, на последовательность стандартных небольших изменений. Это позволяет во многих случаях идентифицировать или указывать, какие из этих стандартных изменений действуют как «узкое место» (или, говоря химическим языком, «этап определения скорости») для развития реакции. Именно структура переходного состояния этого узкого места наиболее важна для объяснения или предсказания наблюдаемых особенностей химических реакций. Кроме того, такое разложение может сделать очевидным, где альтернативные пути доступны, а где нет.Например, реакция, которая протекает через стабильный промежуточный продукт, может быть предметом множества перегруппировок или побочных реакций. Кроме того, поскольку отдельные стадии разложения стандартизированы, они позволяют сделать вывод о структурах важных промежуточных продуктов в конкретной реакции, механизм которой предлагается. Это означает, что разделение реакции на этапы может помочь с первой необходимой характеристикой механизма — позволяя сделать вывод о структурах в «критические моменты» трансформации.Один из способов сделать это — определить, какой из этапов механизма является «узким местом», а затем охарактеризовать этот этап как стандартный. Когда есть общая информация о геометрии переходного состояния этого стандартного типа стадии реакции, эта информация может быть применена к конкретному случаю, чтобы произвести правдоподобные предсказания геометрии соответствующего переходного состояния. Например, если механизм реакции включал стадию согласованного нуклеофильного замещения, то можно было бы правдоподобно сделать вывод, что переходное состояние будет включать приближение нуклеофила и уходящую группу, выходящую с противоположных сторон примерно тригонального плоского центрального атома.Учитывая эту приблизительную геометрию, химик-органик сразу же знает некоторые структурные особенности, которые будут иметь важное влияние на наблюдаемые особенности реакции, такие как — в данном случае — стерические затруднения приближающегося нуклеофила и т. Д. Стандартизованное ступенчатое разложение химического превращения, таким образом, значительно облегчает объяснения и предсказания, которые обычно делают химики-органики. Во многих случаях тонкой характеристики механизма реакции такого рода достаточно, чтобы объяснить все относящиеся к делу экспериментальные факты или провести различие между конкурирующими объяснениями механизма реакции.
Некоторое подтверждение идеи о том, что разложение химической реакции на стандартизованные стадии передает большую часть информации о механизме реакции, которая важна для химиков-органиков, можно найти, рассматривая системы номенклатуры, которые были разработаны химиками-органиками для описания механизмов реакции. Ингольд был источником первой такой системы (см. [Ingold, 1953, главы 7 и 8]), целью которой было различать различные виды реакций замещения и элиминирования.Последующие химики расширили и улучшили исходную систему Ингольда. Текущая система, рекомендованная ИЮПАК (руководящим органом химической номенклатуры), направлена на то, чтобы назвать механизмы в соответствии с «основной валютой молекулярных изменений: образование и разрыв связей» [Guthrie, 1989, p, 25]. Для того чтобы различать механизмы в этих терминах, необходимо передать три характеристики механизма по его названию:
- 1.
Число шагов реакции.
- 2.
Последовательность шагов.
- 3.
Природа этих шагов, включая значительные диффузионные шаги.
[Guthrie, 1989, p, 25]
Стандартные устройства используются для разделения отдельных шагов механизма и записи их порядка (таким образом, отдельные шаги разделяются знаками «+» и пишутся слева по верно). Характер шагов передается путем следования серии правил, которые используют ранее существовавшие схемы для классификации реакций и агентов, участвующих в них.Например, стадии классифицируются как замены, добавления, исключения или перегруппировки, а агенты классифицируются как нуклеофилы или электрофилы. Шаг называется, указывая, сколько связей с центральным атомом (ами) образовано и разорвано, и являются ли эти изменения связей нуклеофильными или электрофильными процессами. Электрофильное присоединение к двойной связи с последующим образованием связи между карбокатионом и нуклеофилом будет обозначено: A E + A N .Это название указывает на то, что реакция представляет собой двухэтапный процесс, в котором образуются две связи. Первая связь образуется между одним из стержневых атомов и электрофилом, а следующая связь формируется на отдельном этапе между оставшимся стержневым атомом и нуклеофилом. Конечно, для более сложных реакций должны использоваться дополнительные устройства, но: «даже такие сложные механизмы можно рассматривать как последовательности легко символизируемых компонентных процессов» [Guthrie, 1989, p. 25]. Теперь я представлю пример того, как механизм химической реакции или класс химических реакций развивается на основе экспериментальных данных.
Теория атомов в молекулах: Введение
Теория атомов в молекулах: ВведениеВведение
Гипотеза молекулярной структуры — что молекула — это совокупность атомов, связанных сетью связей — была выкована в тигле экспериментальной химии девятнадцатого века. Он продолжал служить основным средством систематизации и классификации химических наблюдений. Сложность этой гипотезы заключалась в том, что она не имела прямого отношения к квантовой механике, физике, которая управляет движениями ядер и электронов, составляющих атомы и связи.Действительно, существовало и до сих пор остается преобладающее мнение о том, что эти фундаментальные концепции, будучи несомненно полезными, выходят за рамки теоретического определения. В химии у нас есть понимание, основанное на схеме классификации, которая является одновременно мощной и в то же время ограниченной в силу своей эмпирической природы.
Ричард Фейнман и Джулиан Швингер дали нам переформулировку физики, которая позволяет ставить и отвечать на вопросы «что такое атом в молекуле и как предсказать его свойства?» Эти вопросы были заданы в моей лаборатории, где было продемонстрировано, что эта новая формулировка физики в применении к наблюдаемой топологии распределения электронного заряда в реальном пространстве дает уникальное разбиение некоторой целостной системы на набор ограниченных пространственных областей.Форма и свойства групп, определенных таким образом, точно воспроизводят характеристики, приписываемые атомам и функциональным группам химии. Устанавливая эту ассоциацию, гипотеза молекулярной структуры освобождается от ее эмпирических ограничений, и вся предсказательная сила квантовой механики может быть включена в результирующую теорию — теорию атомов в молекулах и кристаллах.
Теория восстанавливает центральные рабочие концепции гипотезы молекулярной структуры, концепции функциональной группировки атомов с аддитивным и характерным набором свойств, вместе с определением связей, которые связывают атомы и придают структуру.Таким образом, теория не только дает количественную оценку и физическое понимание существующих концепций химии, но и делает возможными новые приложения теории. Эти новые приложения в конечном итоге позволят выполнять на компьютере, прямо параллельно эксперименту, все, что теперь можно делать в лаборатории, но быстрее и эффективнее, за счет объединения функциональных групп теории.