Теория химического строения — Википедия
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу
[1].
Предпосылки создания теории химического строения
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова
Термин «химическое строение» впервые ввёл А. М. Бутлеров 19 сентября 1861 года в докладе «О химическом строении веществ» на химической секции Съезда немецких естествоиспытателей и врачей в Шпейере (опубликованном в том же году на немецком [3] и в следующем — на русском языках[4]). В том же докладе он заложил основы классической теории химического строения. Главные положения этой теории следующие:
- атомы в молекулах веществ соединены друг с другом согласно их валентности, порядок распределения связей в молекуле называется химическим строением;
- изменение этой последовательности приводит к образованию нового вещества с новыми свойствами;
- свойства веществ зависят не только от их качественного и количественного состава, но и от «химического строения», то есть от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильно влияют друг на друга атомы, непосредственно связанные между собой;
- атомы в молекулах оказывают влияние друг на друга, и это влияние приводит к химическим изменениям поведения атома;
- определить состав и строение химического вещества можно по продуктам химических превращений.
Геометрия молекул
В 1864 году Бутлеров первым объясняет явление изомерии, показав что изомеры — это соединения, обладающие одинаковым элементным составом, но различным химическим строением. В 1874 году возникает стереохимия, или трёхмерная структурная химия в форме постулата Вант-Гоффа о тетраэдрической системе валентностей у атома углерода.
В настоящее время принято различать структурную и пространственную изомерию.
Структурную изомерию подразделяют на изомерию скелета, обусловленную различным порядком связи атомов, образующих скелет молекулы, например в Н-бутане и изобутане, и на изомерию положения одинаковых функциональных групп при одинаковом углеродном скелете молекулы, например в орто- , мета- и пара- изомерах ароматических соединений.
Пространственная изомерия обусловлена существованием стереоизомеров, соединений, имеющих одинаковый порядок связей атомов, но различное пространственное расположение. К видам пространственной изомерии относятся: оптическая изомерия, обусловливающая существование энантиомеров — пары стереоизомеров, представляющих собой зеркальные отражения друг от друга, не совмещаемые в пространстве; диастереомерия, обусловливающая существование изомеров, не являющихся энантиомерами; геометрическая изомерия, обусловливающая цис- и транс- изомеров, свойственных соединениям с двойными связями и малыми циклами.
Электронные интерпретации строения молекул
Строение молекулы H2 по БоруС открытием в 1897 году электрона (Дж. Томсоном, Э. Вихертом) появились электронные интерпретации строения молекул. Американский физикохимик Г. Льюис в 1912 году предложил электронную теорию химической связи, по которой связь между атомами в молекуле осуществляется обобществлённой электронной парой. Электронная теория химической связи Льюиса стала основой классической теории строения в органической химии, базирующейся на представлении о парной связи между атомами, образованной дублетом электронов. Валентный штрих между символами элементов в молекуле был заменён двумя точками, обозначающими связывающую электронную пару.
В 1913 году датский физик Нильс Бор предложил рассматривать электронную пару в форме вращающегося кольца, плоскость которого перпендикулярна оси молекулы и равноудалена от ядер атомов.
Динамическое равновесие молекулярной системы, по Бору, достигается за счёт баланса сил между силами взаимного отталкивания ядер и силами притяжения ядер к плоскости кольца электронов. Боровская интерпретация строения молекулы учитывала кулоновское отталкивание электронов, в кольце они находятся в диаметрально противоположном положении.
Молекулу метана Нильс Бор описывал следующим образом:
Ядро углерода, заключённое в очень маленькое кольцо из двух электронов, расположено в центре (тетраэдра), а ядро водорода по углам. Химические связи представляют собой четыре двухэлектронных кольца, вращающихся вокруг линий, соединяющих центр с углами [5].
Квантовая природа межатомных сил
Схема диполь-дипольного взаимодействия двух атомов водорода по ЛондонуВ своём докладе, прочитанном в Королевском колледже в Лондоне в ознаменовании столетия опубликования трудов Дж. Максвелла по электромагнитному излучению, К. Коулсон дал анализ происхождения и сущности межатомных сил, приводящих к образованию молекулы[6]. Коулсон, ссылаясь на работу Лондона, на примере двух атомов водорода показывает «каким образом два нейтральных атома или молекулы могут оказывать притяжение друг к другу на значительном расстоянии».
Ядра A и B двух атомов водорода находятся на расстоянии r друг от друга (рис.). Каждый атом несёт по одном электрону (в P и Q соответственно). Совокупность зарядов +e в A и -e в P приблизительно эквивалентна электрическому диполю, имеющему величину e·AP. Подобным же образом совокупность +e в B и -e в Q приблизительно эквивалентна электрическому диполю e·BQ. Эти два диполя взаимодействуют друг с другом. Общая потенциальная энергия двух диполей, m и m’, находящихся на расстоянии r, равна:
mm′r3−3(mr)(m′r)r5{\displaystyle {\frac {mm’}{r^{3}}}-3{\frac {(mr)(m’r)}{r^{5}}}}[6]
На языке волновой механики это выражение рассматривается как возмущение, действующее на оба атома.
Природа межатомных сил имеет электромагнитный характер и называется диполь-дипольным взаимодействием. Существуют диполь-квадрупольные, квадруполь-квадрупольные и другие взаимодействия, при которых энергия изменяется в зависимости от более высоких степеней 1/r[6].
Распределение электронной плотности в химических соединениях
Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода. Рис. Модель молекулы водорода и её осевая проекцияЭлектронная плотность — это плотность вероятности обнаружения электрона в данной точке пространства. Электронная плотность нормирована и, соответственно, указывает на вероятное число электронов в данном элементарном объёме.
Вероятностную (статистическую) интерпретацию волновой функции сформулировал М. Борн в 1926 году как только было опубликована волновое уравнение Шрёдингера. В 1954 году М. Борн удостоен Нобелевской премии по физике с формулировкой «За фундаментальное исследование в области квантовой механики, особенно за статистическую интерпретацию волновой функции».
Расчёт электронной плотности проводят с использованием уравнения Шрёдингера, которое решается аналитически точно для систем, содержащих только один электрон[7].
Получаемая радиальная функция распределения вероятности нахождения электрона в атоме водорода обладает максимумом при α0, как показано на рисунке. Этот наиболее вероятный радиус совпадает с боровским радиусом и принят в качестве атомной единицы для линейных размеров 1 а. е. (бор) = 0,529177·10−10м ≈ 0,529 Å. Более размытое облако плотности вероятности, полученное при решении уравнения Шрёдингера для атома водорода, значительно отличается от боровской модели атома и согласуется с принципами неопределённости Гейзенберга. С учётом статистической интерпретации волновой функции М. Борна и принципа неопределённости Гейзенберга длины диполей AP и BQ взаимодействующих атомов в расчётах Ф. Лондона достаточно размыты. Размыто и электронное кольцо в модели молекулы водорода по Н. Бору до состояния тороидального электронного облака с неопределёнными границами.
Наиболее вероятный радиус электронного кольца (тора) молекулы водорода (re) определяется боровским радиусом (α0) и длиной химической связи (d): re2 = α02 — (d/2)2; re = 0,374 Å.
Благодаря пространственной симметрии дипольный момент молекулы водорода равен нулю, что соответствует её низкой химической активности[8]. Пространственная симметрия электронной плотности сохраняется, если соединяющиеся в молекулу атомы имеют одинаковую энергию ионизации. В этом случае связывающее электронное кольцо располагается на равном расстоянии от каждого из ядер. Если же потенциалы ионизации различны электронная плотность смещается в сторону атома с большим первым потенциалом ионизации[9]. Смещение электронной плотности приводит к асимметрии распределения электрических зарядов в молекуле, молекула становится полярной системой с определённым дипольным моментом.
Приближённые квантовохимические методы расчёта
Поскольку точное решение уравнения Шрёдингера для атомно-молекулярных систем, содержащих два и более электрона, невозможно, предложены приближённые методы расчёта электронной плотности. Все они возникли в 1930-х годах, проделали значительный путь развития и долгое время дополняли друг друга. Наиболее важные из них — теория валентных связей, теория молекулярных орбиталей, теория кристаллического поля, теория функционала плотности.
В рамках теории валентных связей разработана концепция резонанса (Л. Полинг) и родственная ей концепция мезомерии (К. Ингольд). Концепция резонанса рассмотрена на примере молекулярного иона водорода. Л. Полинг показал, что стабильность молекулярного иона водорода:
вызвана резонансом электрона, то есть движением его «взад и вперёд» между двумя ядрами с «резонансной частотой», равной по величине энергии резонанса (50 ккал/моль), делённой на константу Планка h. Для молекулярного иона в основном состоянии эта частота равна 7.1014сек−1[10].
Концепция резонанса дополняла постулаты классической теории химического строения и утверждала, что если для данного соединения классическая теория допускает построение нескольких приемлемых структурных формул, то действительному состоянию молекул этого соединения отвечает не какая-либо одна отдельная формула, а некоторое их сочетание (наложение, резонанс структур)[11].
Мезомерия является теорией электронного строения химических соединений, согласно которой истинное распределение электронной плотности в молекуле является промежуточным между распределениями, представленными несколькими классическими формулами[12].
Обычно рассматривают положительный и отрицательный мезомерные эффекты:
Винилхлорид: +М-эффект
Аллил-катион: −М-эффект
Кампания идеологического вмешательства в теорию химического строения
Кампания началась в 1949 году с публикации статьи В. М. Татевского и М. М. Шахпаронова «Об одной махистской теории в химии и её пропагандистах»[13]. В качестве главного объекта нападения была выбрана теория резонанса Л. Полинга. Было объявлено, что «представления о реальной молекуле как о чём-то среднем между двумя (и более) крайними абстрактными структурами являются буржуазными и поэтому направленными против всего самого „святого“». Были указаны и пропагандисты теории Я. К. Сыркин и М. Е. Дяткина — авторы книги «Химическая связь и строение молекул», в которой нашла отражение теория резонанса.
В воздухе очередной раз запахло инквизицией. В этой тревожной обстановке ведущие химики страны собрались на Всесоюзное совещание по проблемам химического строения (1951 г., Москва). Стенограмма этого совещания — один из наиболее позорных документов, когда-либо созданных коллективными усилиями ученых, хранится в химических библиотеках всего мира, и от этого срама бог весть когда ещё удастся отмыться… До крови не дошло — спасла оттепель, начавшаяся весной следующего года. Я. К. Сыркин и М. Е. Дяткина, подготовленные недавними друзьями и коллегами к выдаче в качестве первых козлов отпущения, уцелели; более того Я. К. Сыркин в дальнейшем стал академиком[14].
Лайнус Полинг удостоен в 1954 году Нобелевской премии по химии «за исследование природы химической связи и её применение для определения структуры сложных соединений».
Однако консенсус в теории химического строения не был достигнут. В. М. Татевский в курсе «Строение молекул» (1977 г.) отмечал:
… полностью выпадают и «висят в воздухе» представления уходящих в прошлое, но всё ещё фигурирующих в литературе так называемых «теории резонанса» и «теории мезомерии», которые не имеют основы ни в классической теории химического строения, ни в законных приложениях классической физики к вопросам строения молекул, ни в квантовой механике[15].
Лишь в 1991 году проведён принципиальный анализ кампании по борьбе с теорией резонанса и было показано, что эта кампания «нанесла серьёзный ущерб престижу советской науки»[16].
Атомы в молекулах
В классической теории химического строения понятие атома в молекуле является изначальным. Интуитивно ясно, что атом в молекуле меняется, меняются и его свойства в зависимости от окружения этого атома, прежде всего ближайшего. Основным показателем является расстояние между атомами в молекуле, определяющее как длину химической связи, так и прочность химической связи.
В квантовой теории понятие атома вторично. Так, по утверждению В. М. Татевского, молекула не состоит из атомов: «С современной точки зрения ясно, что при образовании молекулы сохраняются не атомы, а только ядра атомов и электроны»[17]
.Было предпринято множество попыток сохранить понятие атома в молекуле, но, тем не менее, практически всегда они не удовлетворяли последующих исследователей по тем или иным причинам.
Одна из наиболее удачных попыток сохранения классической концепции атома в молекуле принадлежит Р. Бейдеру и его сотрудникам[18]. В рамках этой теории (QTAIM) электронная плотность «задаёт некоторое скалярное поле в трёхмерном пространстве, которое может быть охарактеризовано, например, его совокупностью экстремальных точек, линий и поверхностей, особых точек и т. п.»[19].
Таким образом, в квантовой теории атомов в молекулах Р. Бейдера оказывается возможным физическое обоснование ключевых понятий химии «атом», «молекула», «химическая связь» в терминах топологии функции электронной плотности в трёхмерном пространстве и описание химического строения молекул.
Электронная корреляция и конфигурация молекул
Электронная корреляция (взаимная обусловленность движения всех электронов атомной или молекулярной системы как целого определяется электростатическим отталкиванием электронов и статическим особенностями систем, в частности принципом Паули (фермиевская корреляция). Полный учёт электронной корреляции при расчёте энергии и определении электронной структуры системы достигается методом конфигурационного взаимодействия.
Простая и надёжная система правил для понимания и предсказания конфигурации молекул заложена в теории отталкивания электронных пар, наиболее важное правило которой достаточно эмпирично, хотя имеет квантовомеханическое обоснование, заключающееся в принципе Паули, а именно «электронные пары принимают такое расположение на валентной оболочке атома, при котором они максимально удалены друг от друга». При этом конфигурация молекулы будет определяться числом связывающих и неподелённых электронных пар у центрального атома:
- две связывающие электронные пары дают линейную конфигурацию молекулы;
- три — конфигурацию правильного треугольника;
- четыре — конфигурацию тетраэдра;
- пять — тригонально-бипирамидальную конфигурацию;
- шесть — конфигурацию октаэдра.
Наличие неподелённых электронных пар у центрального атома приводит к расширению типов конфигураций молекулы[20].
Строение соединений благородных газов
Молекула дифторида ксенона Молекула тетрафторида ксенона Молекула гексафторида ксенонаОткрытие Н. Бартлеттом в 1962 году первого соединения ксенона положило начало интенсивному развитию химии благородных газов, что привело к получению большого ряда соединений инертных газов и установлению их химического строения.
Оказалось, что все соединения благородных газов имеют обыкновенные ковалентные связи и общепринятые конфигурации молекул. Так, конфигурация молекулы дифторида ксенона линейна: связи ксенон—фтор являются аксиальными и взаимодействуют с тремя неподелёнными электронными парами, находящимися в экваториальных положениях.
В молекуле тетрафторида ксенона осуществляется конфигурация с плоским квадратным расположением лигандов. Неподелённые электронные пары ксенона максимально удалены друг от друга и расположены по разные стороны от этого квадрата.
Структура молекулы гексафторида ксенона представляет собой искажённый октаэдр. «Отклонение атомов фтора от вершин правильного октаэдра указывает на то, что неподелённая электронная пара занимает положение над центром одной из граней октаэдра, а атомы фтора, расположенные в углах этой грани, вынуждены раздвинуться»[20].
Открытие Бартлетта показало ошибочность популярного в то время правила октета,[как?] согласно которому при образовании молекул атомы удовлетворяют свою потребность в достижении 8-электронной валентной оболочки, подобной электронной конфигурации благородных газов за счёт попарного обобществления своих валентных электронов. Оказалось, что атомы благородных газов, имея полностью заполненную валентную оболочку, могут вступать в химические реакции и участвовать в химическом строении молекул.
Строение электронодефицитных соединений
Структурная проблема, связанная с электронодефицитными соединениями, довольно сложна. Фундаментальная трудность заключалась в том, что в молекулах нет достаточного числа валентных электронов для того, чтобы связать все атомы обычными двухэлектронными связями. Так, например, в молекуле диборана имеется двенадцать валентных электронов, все двенадцать нужны для образования ковалентных связей шести атомов водорода с бором, так что для связи атомов бора между собой электронов не остаётся. Сам Полинг допускал, что в диборане функционируют одноэлектронные связи, а молекула в основном состоянии резонирует между семью структурами льюисовского типа, а также между многочисленными структурами, содержащими одноэлектронные связи.
Однако заслуженное признание получили исследования природы химической связи в бороводородах, выполненные американским физикохимиком У. Липскомбом. В его интерпретации в диборане имеют место четыре двух- и две трёхцентровые связи.
Четыре концевые двухцентровые двухэлектронные связи HB лежат в одной плоскости. Два же центральных атома водорода расположены симметрично над этой плоскостью и под нею и объединены с атомами бора двумя трёхцентровыми связями.
В 1976 году Липскомб удостоен Нобелевской премии по химии с формулировкой «за исследование структуры боранов (боргидритов), проясняющих проблему химических связей».
Особенности строения сэндвичевых соединений
Молекула ферроценаДальнейшее развитие теории химического строения связано с открытием и установлением структуры ферроцена[21]. Оказалось, что при взаимодействии хлорида двухвалентного железа и циклопентадиена вместо ожидаемого соединения с двумя химическими связями железо-углерод образуется сэндвичеобразное соединение, в котором все 10 атомов углерода соединены с атомом железа.
Рентгеноструктурными исследованиями Э. Фишера установлено, что все атомы углерода в молекуле ферроцена структурно и химически эквивалентны. Атом металла взаимодействует не с одним или двумя конкретными атомами углерода, а со всеми атомами углерода двух циклопентадиенильных колец сразу. Атом металла как бы завис в пространстве между двумя циклами, представленных параллельными правильными пятиугольниками.
В настоящее время получены металлоцены — бициклопентадиенильные соединения переходных металлов для большинства d-элементов.
Сэндвичевые соединения образуются также с использованием в качестве органического фрагмента бензола или циклооктатетраена. Так, например, в ураноцене (см. рисунок) атом урана удерживает два восьмичленных кольца. Все 16 связей уран—углерод в ураноцене идентичны.
В 1973 году Э. Фишер и Дж. Уилкинсон удостоены Нобелевской премии по химии с формулировкой «За новаторскую, проделанную независимо друг от друга, работу в области металлорганических, так называемых сэндвичевых соединений».
Считается, что в сэндвичевых соединениях реализуется многоцентровая связь, тип химической связи, в которой в отличие от двухцентровой двухэлектронной связи, связывающие электронные пары распределены в пространстве трех или большего числа атомных центров молекулы, иона, радикала.[22]
Примечания
- ↑ 1 2 Совещание по теории химического строения в органической химии. Успехи физических наук.. — 1951, октябрь. — Т. XLV, вып. 2. — С. 277—293.
- ↑ Химический энциклопедический словарь / гл. ред. И. Л. Кнунянц. — Сов. энциклопедия, 1983. — С. 652. — 792 с.
- ↑ Butlerow A. (1861). «Einiges über die chemische Structur der Körper. (Vorgetragen in der chemischen Section der 36. Versammlung deutscher Naturforscher und Aerzte zu Speyer am 19.Septbr.)». Zeitschrift für Chemie und Pharmacie 4: 549-560.
- ↑ Бутлеров А. О химическом строении веществ // Учёные записки Казанского университета (отд. физ.-мат. и мед. наук). Вып.1, отд.1. — 1862. — С. 1—11.
- ↑ Бор Н. Избранные научные труды. — М.: «Наука», 1970. — Т. 1. — С. 147. — 584 с.
- ↑ 1 2 3 Коулсон К. Успехи физических наук (статья «Межатомные силы — от Максвелла до Шрёдингера»). — 1963, ноябрь. — Т. LXXXI, вып.3. — С. 545-556.
- ↑ Шусторович Е. М. Химическая связь. Сущность и проблемы.. — М.: «Наука», 1973. — С. 6. — 232 с.
- ↑ Потапов А. А. Ренессанс классического атома. — М.: «Наука», 2011. — С. 352. — 444 с.
- ↑ Ганкин В. Ю., Ганкин Ю. В. Как образуется химическая связь и протекают химические реакции. — М.: «Граница». — С. 65. — 320 с.
- ↑ Паулинг Л. Природа химической связи. — М.-Л.: Химическая литература, 1947. — С. 25—26. — 440 с.
- ↑ Новая иллюстрированная энциклопедия. Пр-Ро. Кн.15. — М.: Большая российская энциклопедия, 2002. — С. 169. — 255 с.
- ↑ Новая иллюстрированная энциклопедия. Ма-Мо. Кн.11. — М.: Большая российская энциклопедия, 2002. — С. 141. — 255 с.
- ↑ Вопросы философии. — 1949. — № 3. — С. 176—192.
- ↑ Охлобыстин О. Ю. Жизнь и смерть химических идей. Очерки по истории теоретической химии.. — М.: Наука, 1989. — С. 184. — 190 с.
- ↑ Татевский В. М. Строение молекул.. — М.: Химия, 1977. — С. 10. — 512 с.
- ↑ Сонин А. С. Печальный юбилей одной кампании. (рус.) // Вестник РАН. — 1991. — Т. 61, № 8. — С. 96—107. Архивировано 5 мая 2010 года.
- ↑ Татевский В. М. Квантовая механика и теория строения молекул. — М.: Издательство МГУ, 1965. — С. 17. — 162 с.
- ↑ Бейдер Р. Атомы в молекулах. Квантовая теория. — М.: Мир, 2001. — 532 с.
- ↑ Степанов Н. Ф. Квантовая механика и квантовая химия. — М.: Мир, 2001. — С. 487. — 519 с.
- ↑ 1 2 Р. Гиллеспи. Геометрия молекул. — М.: Мир, 1975. — 278 с.
- ↑ Несмеянов А.Н. Ферроцен и родственные соединения. Избранные труды 1969 — 1979. — М.: Советская энциклопедия, 1982.
- ↑ Химический Энциклопедический словарь. — М.: Советская энциклопедия, 1983. — 792 с.
wikipedia.green
Теория химического строения — Википедия. Что такое Теория химического строения
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу[1].
Предпосылки создания теории химического строения
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова
Термин «химическое строение» впервые ввёл А. М. Бутлеров 19 сентября 1861 года в докладе «О химическом строении веществ» на химической секции Съезда немецких естествоиспытателей и врачей в Шпейере (опубликованном в том же году на немецком[3] и в следующем — на русском языках[4]). В том же докладе он заложил основы классической теории химического строения. Главные положения этой теории следующие:
- атомы в молекулах веществ соединены друг с другом согласно их валентности, порядок распределения связей в молекуле называется химическим строением;
- изменение этой последовательности приводит к образованию нового вещества с новыми свойствами;
- свойства веществ зависят не только от их качественного и количественного состава, но и от «химического строения», то есть от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильно влияют друг на друга атомы, непосредственно связанные между собой;
- атомы в молекулах оказывают влияние друг на друга, и это влияние приводит к химическим изменениям поведения атома;
- определить состав и строение химического вещества можно по продуктам химических превращений.
Геометрия молекул
В 1864 году Бутлеров первым объясняет явление изомерии, показав что изомеры — это соединения, обладающие одинаковым элементным составом, но различным химическим строением. В 1874 году возникает стереохимия, или трёхмерная структурная химия в форме постулата Вант-Гоффа о тетраэдрической системе валентностей у атома углерода.
В настоящее время принято различать структурную и пространственную изомерию.
Структурную изомерию подразделяют на изомерию скелета, обусловленную различным порядком связи атомов, образующих скелет молекулы, например в Н-бутане и изобутане, и на изомерию положения одинаковых функциональных групп при одинаковом углеродном скелете молекулы, например в орто- , мета- и пара- изомерах ароматических соединений.
Пространственная изомерия обусловлена существованием стереоизомеров, соединений, имеющих одинаковый порядок связей атомов, но различное пространственное расположение. К видам пространственной изомерии относятся: оптическая изомерия, обусловливающая существование энантиомеров — пары стереоизомеров, представляющих собой зеркальные отражения друг от друга, не совмещаемые в пространстве; диастереомерия, обусловливающая существование изомеров, не являющихся энантиомерами; геометрическая изомерия, обусловливающая цис- и транс- изомеров, свойственных соединениям с двойными связями и малыми циклами.
Электронные интерпретации строения молекул
Строение молекулы H2 по БоруС открытием в 1897 году электрона (Дж. Томсоном, Э. Вихертом) появились электронные интерпретации строения молекул. Американский физикохимик Г. Льюис в 1912 году предложил электронную теорию химической связи, по которой связь между атомами в молекуле осуществляется обобществлённой электронной парой. Электронная теория химической связи Льюиса стала основой классической теории строения в органической химии, базирующейся на представлении о парной связи между атомами, образованной дублетом электронов. Валентный штрих между символами элементов в молекуле был заменён двумя точками, обозначающими связывающую электронную пару.
В 1913 году датский физик Нильс Бор предложил рассматривать электронную пару в форме вращающегося кольца, плоскость которого перпендикулярна оси молекулы и равноудалена от ядер атомов.
Динамическое равновесие молекулярной системы, по Бору, достигается за счёт баланса сил между силами взаимного отталкивания ядер и силами притяжения ядер к плоскости кольца электронов. Боровская интерпретация строения молекулы учитывала кулоновское отталкивание электронов, в кольце они находятся в диаметрально противоположном положении.
Молекулу метана Нильс Бор описывал следующим образом:
Ядро углерода, заключённое в очень маленькое кольцо из двух электронов, расположено в центре (тетраэдра), а ядро водорода по углам. Химические связи представляют собой четыре двухэлектронных кольца, вращающихся вокруг линий, соединяющих центр с углами[5].
Квантовая природа межатомных сил
Схема диполь-дипольного взаимодействия двух атомов водорода по ЛондонуВ своём докладе, прочитанном в Королевском колледже в Лондоне в ознаменовании столетия опубликования трудов Дж. Максвелла по электромагнитному излучению, К. Коулсон дал анализ происхождения и сущности межатомных сил, приводящих к образованию молекулы[6]. Коулсон, ссылаясь на работу Лондона, на примере двух атомов водорода показывает «каким образом два нейтральных атома или молекулы могут оказывать притяжение друг к другу на значительном расстоянии».
Ядра A и B двух атомов водорода находятся на расстоянии r друг от друга (рис.). Каждый атом несёт по одном электрону (в P и Q соответственно). Совокупность зарядов +e в A и -e в P приблизительно эквивалентна электрическому диполю, имеющему величину e·AP. Подобным же образом совокупность +e в B и -e в Q приблизительно эквивалентна электрическому диполю e·BQ. Эти два диполя взаимодействуют друг с другом. Общая потенциальная энергия двух диполей, m и m’, находящихся на расстоянии r, равна:
mm′r3−3(mr)(m′r)r5{\displaystyle {\frac {mm’}{r^{3}}}-3{\frac {(mr)(m’r)}{r^{5}}}}[6]
На языке волновой механики это выражение рассматривается как возмущение, действующее на оба атома.
Природа межатомных сил имеет электромагнитный характер и называется диполь-дипольным взаимодействием. Существуют диполь-квадрупольные, квадруполь-квадрупольные и другие взаимодействия, при которых энергия изменяется в зависимости от более высоких степеней 1/r[6].
Распределение электронной плотности в химических соединениях
 Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода.
Рис. Модель молекулы водорода и её осевая проекция
Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода.
Рис. Модель молекулы водорода и её осевая проекцияЭлектронная плотность — это плотность вероятности обнаружения электрона в данной точке пространства. Электронная плотность нормирована и, соответственно, указывает на вероятное число электронов в данном элементарном объёме.
Вероятностную (статистическую) интерпретацию волновой функции сформулировал М. Борн в 1926 году как только было опубликована волновое уравнение Шрёдингера. В 1954 году М. Борн удостоен Нобелевской премии по физике с формулировкой «За фундаментальное исследование в области квантовой механики, особенно за статистическую интерпретацию волновой функции».
Расчёт электронной плотности проводят с использованием уравнения Шрёдингера, которое решается аналитически точно для систем, содержащих только один электрон[7].
Получаемая радиальная функция распределения вероятности нахождения электрона в атоме водорода обладает максимумом при α0, как показано на рисунке. Этот наиболее вероятный радиус совпадает с боровским радиусом и принят в качестве атомной единицы для линейных размеров 1 а. е. (бор) = 0,529177·10−10м ≈ 0,529 Å. Более размытое облако плотности вероятности, полученное при решении уравнения Шрёдингера для атома водорода, значительно отличается от боровской модели атома и согласуется с принципами неопределённости Гейзенберга. С учётом статистической интерпретации волновой функции М. Борна и принципа неопределённости Гейзенберга длины диполей AP и BQ взаимодействующих атомов в расчётах Ф. Лондона достаточно размыты. Размыто и электронное кольцо в модели молекулы водорода по Н. Бору до состояния тороидального электронного облака с неопределёнными границами.
Наиболее вероятный радиус электронного кольца (тора) молекулы водорода (re) определяется боровским радиусом (α0) и длиной химической связи (d): re2 = α02 — (d/2)2; re = 0,374 Å.
Благодаря пространственной симметрии дипольный момент молекулы водорода равен нулю, что соответствует её низкой химической активности[8]. Пространственная симметрия электронной плотности сохраняется, если соединяющиеся в молекулу атомы имеют одинаковую энергию ионизации. В этом случае связывающее электронное кольцо располагается на равном расстоянии от каждого из ядер. Если же потенциалы ионизации различны электронная плотность смещается в сторону атома с большим первым потенциалом ионизации[9]. Смещение электронной плотности приводит к асимметрии распределения электрических зарядов в молекуле, молекула становится полярной системой с определённым дипольным моментом.
Приближённые квантовохимические методы расчёта
Поскольку точное решение уравнения Шрёдингера для атомно-молекулярных систем, содержащих два и более электрона, невозможно, предложены приближённые методы расчёта электронной плотности. Все они возникли в 1930-х годах, проделали значительный путь развития и долгое время дополняли друг друга. Наиболее важные из них — теория валентных связей, теория молекулярных орбиталей, теория кристаллического поля, теория функционала плотности.
В рамках теории валентных связей разработана концепция резонанса (Л. Полинг) и родственная ей концепция мезомерии (К. Ингольд). Концепция резонанса рассмотрена на примере молекулярного иона водорода. Л. Полинг показал, что стабильность молекулярного иона водорода:
вызвана резонансом электрона, то есть движением его «взад и вперёд» между двумя ядрами с «резонансной частотой», равной по величине энергии резонанса (50 ккал/моль), делённой на константу Планка h. Для молекулярного иона в основном состоянии эта частота равна 7.1014сек−1[10].
Концепция резонанса дополняла постулаты классической теории химического строения и утверждала, что если для данного соединения классическая теория допускает построение нескольких приемлемых структурных формул, то действительному состоянию молекул этого соединения отвечает не какая-либо одна отдельная формула, а некоторое их сочетание (наложение, резонанс структур)[11].
Мезомерия является теорией электронного строения химических соединений, согласно которой истинное распределение электронной плотности в молекуле является промежуточным между распределениями, представленными несколькими классическими формулами[12].
Обычно рассматривают положительный и отрицательный мезомерные эффекты:
Винилхлорид: +М-эффект
Аллил-катион: −М-эффект
Кампания идеологического вмешательства в теорию химического строения
Кампания началась в 1949 году с публикации статьи В. М. Татевского и М. М. Шахпаронова «Об одной махистской теории в химии и её пропагандистах»[13]. В качестве главного объекта нападения была выбрана теория резонанса Л. Полинга. Было объявлено, что «представления о реальной молекуле как о чём-то среднем между двумя (и более) крайними абстрактными структурами являются буржуазными и поэтому направленными против всего самого „святого“». Были указаны и пропагандисты теории Я. К. Сыркин и М. Е. Дяткина — авторы книги «Химическая связь и строение молекул», в которой нашла отражение теория резонанса.
В воздухе очередной раз запахло инквизицией. В этой тревожной обстановке ведущие химики страны собрались на Всесоюзное совещание по проблемам химического строения (1951 г., Москва). Стенограмма этого совещания — один из наиболее позорных документов, когда-либо созданных коллективными усилиями ученых, хранится в химических библиотеках всего мира, и от этого срама бог весть когда ещё удастся отмыться… До крови не дошло — спасла оттепель, начавшаяся весной следующего года. Я. К. Сыркин и М. Е. Дяткина, подготовленные недавними друзьями и коллегами к выдаче в качестве первых козлов отпущения, уцелели; более того Я. К. Сыркин в дальнейшем стал академиком[14].
Лайнус Полинг удостоен в 1954 году Нобелевской премии по химии «за исследование природы химической связи и её применение для определения структуры сложных соединений».
Однако консенсус в теории химического строения не был достигнут. В. М. Татевский в курсе «Строение молекул» (1977 г.) отмечал:
… полностью выпадают и «висят в воздухе» представления уходящих в прошлое, но всё ещё фигурирующих в литературе так называемых «теории резонанса» и «теории мезомерии», которые не имеют основы ни в классической теории химического строения, ни в законных приложениях классической физики к вопросам строения молекул, ни в квантовой механике[15].
Лишь в 1991 году проведён принципиальный анализ кампании по борьбе с теорией резонанса и было показано, что эта кампания «нанесла серьёзный ущерб престижу советской науки»[16].
Атомы в молекулах
В классической теории химического строения понятие атома в молекуле является изначальным. Интуитивно ясно, что атом в молекуле меняется, меняются и его свойства в зависимости от окружения этого атома, прежде всего ближайшего. Основным показателем является расстояние между атомами в молекуле, определяющее как длину химической связи, так и прочность химической связи.
В квантовой теории понятие атома вторично. Так, по утверждению В. М. Татевского, молекула не состоит из атомов: «С современной точки зрения ясно, что при образовании молекулы сохраняются не атомы, а только ядра атомов и электроны»[17].
Было предпринято множество попыток сохранить понятие атома в молекуле, но, тем не менее, практически всегда они не удовлетворяли последующих исследователей по тем или иным причинам.
Одна из наиболее удачных попыток сохранения классической концепции атома в молекуле принадлежит Р. Бейдеру и его сотрудникам[18]. В рамках этой теории (QTAIM) электронная плотность «задаёт некоторое скалярное поле в трёхмерном пространстве, которое может быть охарактеризовано, например, его совокупностью экстремальных точек, линий и поверхностей, особых точек и т. п.»[19].
Таким образом, в квантовой теории атомов в молекулах Р. Бейдера оказывается возможным физическое обоснование ключевых понятий химии «атом», «молекула», «химическая связь» в терминах топологии функции электронной плотности в трёхмерном пространстве и описание химического строения молекул.
Электронная корреляция и конфигурация молекул
Электронная корреляция (взаимная обусловленность движения всех электронов атомной или молекулярной системы как целого определяется электростатическим отталкиванием электронов и статическим особенностями систем, в частности принципом Паули (фермиевская корреляция). Полный учёт электронной корреляции при расчёте энергии и определении электронной структуры системы достигается методом конфигурационного взаимодействия.
Простая и надёжная система правил для понимания и предсказания конфигурации молекул заложена в теории отталкивания электронных пар, наиболее важное правило которой достаточно эмпирично, хотя имеет квантовомеханическое обоснование, заключающееся в принципе Паули, а именно «электронные пары принимают такое расположение на валентной оболочке атома, при котором они максимально удалены друг от друга». При этом конфигурация молекулы будет определяться числом связывающих и неподелённых электронных пар у центрального атома:
- две связывающие электронные пары дают линейную конфигурацию молекулы;
- три — конфигурацию правильного треугольника;
- четыре — конфигурацию тетраэдра;
- пять — тригонально-бипирамидальную конфигурацию;
- шесть — конфигурацию октаэдра.
Наличие неподелённых электронных пар у центрального атома приводит к расширению типов конфигураций молекулы[20].
Строение соединений благородных газов
 Молекула дифторида ксенона
Молекула дифторида ксенона 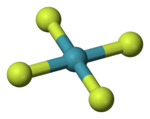 Молекула тетрафторида ксенона
Молекула тетрафторида ксенона  Молекула гексафторида ксенона
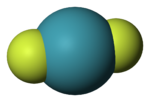
Молекула гексафторида ксенонаОткрытие Н. Бартлеттом в 1962 году первого соединения ксенона положило начало интенсивному развитию химии благородных газов, что привело к получению большого ряда соединений инертных газов и установлению их химического строения.
Оказалось, что все соединения благородных газов имеют обыкновенные ковалентные связи и общепринятые конфигурации молекул. Так, конфигурация молекулы дифторида ксенона линейна: связи ксенон—фтор являются аксиальными и взаимодействуют с тремя неподелёнными электронными парами, находящимися в экваториальных положениях.
В молекуле тетрафторида ксенона осуществляется конфигурация с плоским квадратным расположением лигандов. Неподелённые электронные пары ксенона максимально удалены друг от друга и расположены по разные стороны от этого квадрата.
Структура молекулы гексафторида ксенона представляет собой искажённый октаэдр. «Отклонение атомов фтора от вершин правильного октаэдра указывает на то, что неподелённая электронная пара занимает положение над центром одной из граней октаэдра, а атомы фтора, расположенные в углах этой грани, вынуждены раздвинуться»[20].
Открытие Бартлетта показало ошибочность популярного в то время правила октета,[как?] согласно которому при образовании молекул атомы удовлетворяют свою потребность в достижении 8-электронной валентной оболочки, подобной электронной конфигурации благородных газов за счёт попарного обобществления своих валентных электронов. Оказалось, что атомы благородных газов, имея полностью заполненную валентную оболочку, могут вступать в химические реакции и участвовать в химическом строении молекул.
Строение электронодефицитных соединений
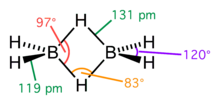
Структурная проблема, связанная с электронодефицитными соединениями, довольно сложна. Фундаментальная трудность заключалась в том, что в молекулах нет достаточного числа валентных электронов для того, чтобы связать все атомы обычными двухэлектронными связями. Так, например, в молекуле диборана имеется двенадцать валентных электронов, все двенадцать нужны для образования ковалентных связей шести атомов водорода с бором, так что для связи атомов бора между собой электронов не остаётся. Сам Полинг допускал, что в диборане функционируют одноэлектронные связи, а молекула в основном состоянии резонирует между семью структурами льюисовского типа, а также между многочисленными структурами, содержащими одноэлектронные связи.
Однако заслуженное признание получили исследования природы химической связи в бороводородах, выполненные американским физикохимиком У. Липскомбом. В его интерпретации в диборане имеют место четыре двух- и две трёхцентровые связи.
Четыре концевые двухцентровые двухэлектронные связи HB лежат в одной плоскости. Два же центральных атома водорода расположены симметрично над этой плоскостью и под нею и объединены с атомами бора двумя трёхцентровыми связями.
В 1976 году Липскомб удостоен Нобелевской премии по химии с формулировкой «за исследование структуры боранов (боргидритов), проясняющих проблему химических связей».
Особенности строения сэндвичевых соединений
Молекула ферроцена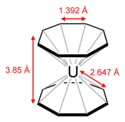
Дальнейшее развитие теории химического строения связано с открытием и установлением структуры ферроцена[21]. Оказалось, что при взаимодействии хлорида двухвалентного железа и циклопентадиена вместо ожидаемого соединения с двумя химическими связями железо-углерод образуется сэндвичеобразное соединение, в котором все 10 атомов углерода соединены с атомом железа.
Рентгеноструктурными исследованиями Э. Фишера установлено, что все атомы углерода в молекуле ферроцена структурно и химически эквивалентны. Атом металла взаимодействует не с одним или двумя конкретными атомами углерода, а со всеми атомами углерода двух циклопентадиенильных колец сразу. Атом металла как бы завис в пространстве между двумя циклами, представленных параллельными правильными пятиугольниками.
В настоящее время получены металлоцены — бициклопентадиенильные соединения переходных металлов для большинства d-элементов.
Сэндвичевые соединения образуются также с использованием в качестве органического фрагмента бензола или циклооктатетраена. Так, например, в ураноцене (см. рисунок) атом урана удерживает два восьмичленных кольца. Все 16 связей уран—углерод в ураноцене идентичны.
В 1973 году Э. Фишер и Дж. Уилкинсон удостоены Нобелевской премии по химии с формулировкой «За новаторскую, проделанную независимо друг от друга, работу в области металлорганических, так называемых сэндвичевых соединений».
Считается, что в сэндвичевых соединениях реализуется многоцентровая связь, тип химической связи, в которой в отличие от двухцентровой двухэлектронной связи, связывающие электронные пары распределены в пространстве трех или большего числа атомных центров молекулы, иона, радикала.[22]
Примечания
- ↑ 1 2 Совещание по теории химического строения в органической химии. Успехи физических наук.. — 1951, октябрь. — Т. XLV, вып. 2. — С. 277—293.
- ↑ Химический энциклопедический словарь / гл. ред. И. Л. Кнунянц. — Сов. энциклопедия, 1983. — С. 652. — 792 с.
- ↑ Butlerow A. (1861). «Einiges über die chemische Structur der Körper. (Vorgetragen in der chemischen Section der 36. Versammlung deutscher Naturforscher und Aerzte zu Speyer am 19.Septbr.)». Zeitschrift für Chemie und Pharmacie 4: 549-560.
- ↑ Бутлеров А. О химическом строении веществ // Учёные записки Казанского университета (отд. физ.-мат. и мед. наук). Вып.1, отд.1. — 1862. — С. 1—11.
- ↑ Бор Н. Избранные научные труды. — М.: «Наука», 1970. — Т. 1. — С. 147. — 584 с.
- ↑ 1 2 3 Коулсон К. Успехи физических наук (статья «Межатомные силы — от Максвелла до Шрёдингера»). — 1963, ноябрь. — Т. LXXXI, вып.3. — С. 545-556.
- ↑ Шусторович Е. М. Химическая связь. Сущность и проблемы.. — М.: «Наука», 1973. — С. 6. — 232 с.
- ↑ Потапов А. А. Ренессанс классического атома. — М.: «Наука», 2011. — С. 352. — 444 с.
- ↑ Ганкин В. Ю., Ганкин Ю. В. Как образуется химическая связь и протекают химические реакции. — М.: «Граница». — С. 65. — 320 с.
- ↑ Паулинг Л. Природа химической связи. — М.-Л.: Химическая литература, 1947. — С. 25—26. — 440 с.
- ↑ Новая иллюстрированная энциклопедия. Пр-Ро. Кн.15. — М.: Большая российская энциклопедия, 2002. — С. 169. — 255 с.
- ↑ Новая иллюстрированная энциклопедия. Ма-Мо. Кн.11. — М.: Большая российская энциклопедия, 2002. — С. 141. — 255 с.
- ↑ Вопросы философии. — 1949. — № 3. — С. 176—192.
- ↑ Охлобыстин О. Ю. Жизнь и смерть химических идей. Очерки по истории теоретической химии.. — М.: Наука, 1989. — С. 184. — 190 с.
- ↑ Татевский В. М. Строение молекул.. — М.: Химия, 1977. — С. 10. — 512 с.
- ↑ Сонин А. С. Печальный юбилей одной кампании. (рус.) // Вестник РАН. — 1991. — Т. 61, № 8. — С. 96—107. Архивировано 5 мая 2010 года.
- ↑ Татевский В. М. Квантовая механика и теория строения молекул. — М.: Издательство МГУ, 1965. — С. 17. — 162 с.
- ↑ Бейдер Р. Атомы в молекулах. Квантовая теория. — М.: Мир, 2001. — 532 с.
- ↑ Степанов Н. Ф. Квантовая механика и квантовая химия. — М.: Мир, 2001. — С. 487. — 519 с.
- ↑ 1 2 Р. Гиллеспи. Геометрия молекул. — М.: Мир, 1975. — 278 с.
- ↑ Несмеянов А.Н. Ферроцен и родственные соединения. Избранные труды 1969 — 1979. — М.: Советская энциклопедия, 1982.
- ↑ Химический Энциклопедический словарь. — М.: Советская энциклопедия, 1983. — 792 с.
wiki.sc
Теория химического строения Википедия
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу[1].
Предпосылки создания теории химического строения[ | ]
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова[ |
ru-wiki.ru
Теория химического строения Википедия
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу[1].
Предпосылки создания теории химического строения
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова
Термин «химическое строение» впервые ввёл А. М. Бутлеров 19 сентября 1861 года в докладе «О химическом строении веществ» на химической секции Съезда немецких естествоиспытателей и врачей в Шпейере (опубликованном в том же году на немецком[3] и в следующем — на русском языках[4]). В том же докладе он заложил основы классической теории химического строения. Главные положения этой теории следующие:
- атомы в молекулах веществ соединены друг с другом согласно их валентности, порядок распределения связей в молекуле называется химическим строением;
- изменение этой последовательности приводит к образованию нового вещества с новыми свойствами;
- свойства веществ зависят не только от их качественного и количественного состава, но и от «химического строения», то есть от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильно влияют друг на друга атомы, непосредственно связанные между собой;
- атомы в молекулах оказывают влияние друг на друга, и это влияние приводит к химическим изменениям поведения атома;
- определить состав и строение химического вещества можно по продуктам химических превращений.
Геометрия молекул
В 1864 году Бутлеров первым объясняет явление изомерии, показав что изомеры — это соединения, обладающие одинаковым элементным составом, но различным химическим строением. В 1874 году возникает стереохимия, или трёхмерная структурная химия в форме постулата Вант-Гоффа о тетраэдрической системе валентностей у атома углерода.
В настоящее время принято различать структурную и пространственную изомерию.
Структурную изомерию подразделяют на изомерию скелета, обусловленную различным порядком связи атомов, образующих скелет молекулы, например в Н-бутане и изобутане, и на изомерию положения одинаковых функциональных групп при одинаковом углеродном скелете молекулы, например в орто- , мета- и пара- изомерах ароматических соединений.
Пространственная изомерия обусловлена существование
ruwikiorg.ru
Теория химического строения — WiKi
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу[1].
Предпосылки создания теории химического строения
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова
Термин «химическое строение» впервые ввёл А. М. Бутлеров 19 сентября 1861 года в докладе «О химическом строении веществ» на химической секции Съезда немецких естествоиспытателей и врачей в Шпейере (опубликованном в том же году на немецком[3] и в следующем — на русском языках[4]). В том же докладе он заложил основы классической теории химического строения. Главные положения этой теории следующие:
- атомы в молекулах веществ соединены друг с другом согласно их валентности, порядок распределения связей в молекуле называется химическим строением;
- изменение этой последовательности приводит к образованию нового вещества с новыми свойствами;
- свойства веществ зависят не только от их качественного и количественного состава, но и от «химического строения», то есть от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильно влияют друг на друга атомы, непосредственно связанные между собой;
- атомы в молекулах оказывают влияние друг на друга, и это влияние приводит к химическим изменениям поведения атома;
- определить состав и строение химического вещества можно по продуктам химических превращений.
Геометрия молекул
В 1864 году Бутлеров первым объясняет явление изомерии, показав что изомеры — это соединения, обладающие одинаковым элементным составом, но различным химическим строением. В 1874 году возникает стереохимия, или трёхмерная структурная химия в форме постулата Вант-Гоффа о тетраэдрической системе валентностей у атома углерода.
В настоящее время принято различать структурную и пространственную изомерию.
Структурную изомерию подразделяют на изомерию скелета, обусловленную различным порядком связи атомов, образующих скелет молекулы, например в Н-бутане и изобутане, и на изомерию положения одинаковых функциональных групп при одинаковом углеродном скелете молекулы, например в орто- , мета- и пара- изомерах ароматических соединений.
Пространственная изомерия обусловлена существованием стереоизомеров, соединений, имеющих одинаковый порядок связей атомов, но различное пространственное расположение. К видам пространственной изомерии относятся: оптическая изомерия, обусловливающая существование энантиомеров — пары стереоизомеров, представляющих собой зеркальные отражения друг от друга, не совмещаемые в пространстве; диастереомерия, обусловливающая существование изомеров, не являющихся энантиомерами; геометрическая изомерия, обусловливающая цис- и транс- изомеров, свойственных соединениям с двойными связями и малыми циклами.
Электронные интерпретации строения молекул
Строение молекулы H2 по БоруС открытием в 1897 году электрона (Дж. Томсоном, Э. Вихертом) появились электронные интерпретации строения молекул. Американский физикохимик Г. Льюис в 1912 году предложил электронную теорию химической связи, по которой связь между атомами в молекуле осуществляется обобществлённой электронной парой. Электронная теория химической связи Льюиса стала основой классической теории строения в органической химии, базирующейся на представлении о парной связи между атомами, образованной дублетом электронов. Валентный штрих между символами элементов в молекуле был заменён двумя точками, обозначающими связывающую электронную пару.
В 1913 году датский физик Нильс Бор предложил рассматривать электронную пару в форме вращающегося кольца, плоскость которого перпендикулярна оси молекулы и равноудалена от ядер атомов.
Динамическое равновесие молекулярной системы, по Бору, достигается за счёт баланса сил между силами взаимного отталкивания ядер и силами притяжения ядер к плоскости кольца электронов. Боровская интерпретация строения молекулы учитывала кулоновское отталкивание электронов, в кольце они находятся в диаметрально противоположном положении.
Молекулу метана Нильс Бор описывал следующим образом:
Ядро углерода, заключённое в очень маленькое кольцо из двух электронов, расположено в центре (тетраэдра), а ядро водорода по углам. Химические связи представляют собой четыре двухэлектронных кольца, вращающихся вокруг линий, соединяющих центр с углами[5].
Квантовая природа межатомных сил
Схема диполь-дипольного взаимодействия двух атомов водорода по ЛондонуВ своём докладе, прочитанном в Королевском колледже в Лондоне в ознаменовании столетия опубликования трудов Дж. Максвелла по электромагнитному излучению, К. Коулсон дал анализ происхождения и сущности межатомных сил, приводящих к образованию молекулы[6]. Коулсон, ссылаясь на работу Лондона, на примере двух атомов водорода показывает «каким образом два нейтральных атома или молекулы могут оказывать притяжение друг к другу на значительном расстоянии».
Ядра A и B двух атомов водорода находятся на расстоянии r друг от друга (рис.). Каждый атом несёт по одном электрону (в P и Q соответственно). Совокупность зарядов +e в A и -e в P приблизительно эквивалентна электрическому диполю, имеющему величину e·AP. Подобным же образом совокупность +e в B и -e в Q приблизительно эквивалентна электрическому диполю e·BQ. Эти два диполя взаимодействуют друг с другом. Общая потенциальная энергия двух диполей, m и m’, находящихся на расстоянии r, равна:
mm′r3−3(mr)(m′r)r5{\displaystyle {\frac {mm’}{r^{3}}}-3{\frac {(mr)(m’r)}{r^{5}}}} [6]
На языке волновой механики это выражение рассматривается как возмущение, действующее на оба атома.
Природа межатомных сил имеет электромагнитный характер и называется диполь-дипольным взаимодействием. Существуют диполь-квадрупольные, квадруполь-квадрупольные и другие взаимодействия, при которых энергия изменяется в зависимости от более высоких степеней 1/r[6].
Распределение электронной плотности в химических соединениях
Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода. Рис. Модель молекулы водорода и её осевая проекцияЭлектронная плотность — это плотность вероятности обнаружения электрона в данной точке пространства. Электронная плотность нормирована и, соответственно, указывает на вероятное число электронов в данном элементарном объёме.
Вероятностную (статистическую) интерпретацию волновой функции сформулировал М. Борн в 1926 году как только было опубликована волновое уравнение Шрёдингера. В 1954 году М. Борн удостоен Нобелевской премии по физике с формулировкой «За фундаментальное исследование в области квантовой механики, особенно за статистическую интерпретацию волновой функции».
Расчёт электронной плотности проводят с использованием уравнения Шрёдингера, которое решается аналитически точно для систем, содержащих только один электрон[7].
Получаемая радиальная функция распределения вероятности нахождения электрона в атоме водорода обладает максимумом при α0, как показано на рисунке. Этот наиболее вероятный радиус совпадает с боровским радиусом и принят в качестве атомной единицы для линейных размеров 1 а. е. (бор) = 0,529177·10−10м ≈ 0,529 Å. Более размытое облако плотности вероятности, полученное при решении уравнения Шрёдингера для атома водорода, значительно отличается от боровской модели атома и согласуется с принципами неопределённости Гейзенберга. С учётом статистической интерпретации волновой функции М. Борна и принципа неопределённости Гейзенберга длины диполей AP и BQ взаимодействующих атомов в расчётах Ф. Лондона достаточно размыты. Размыто и электронное кольцо в модели молекулы водорода по Н. Бору до состояния тороидального электронного облака с неопределёнными границами.
Наиболее вероятный радиус электронного кольца (тора) молекулы водорода (re) определяется боровским радиусом (α0) и длиной химической связи (d): re2 = α02 — (d/2)2; re = 0,374 Å.
Благодаря пространственной симметрии дипольный момент молекулы водорода равен нулю, что соответствует её низкой химической активности[8]. Пространственная симметрия электронной плотности сохраняется, если соединяющиеся в молекулу атомы имеют одинаковую энергию ионизации. В этом случае связывающее электронное кольцо располагается на равном расстоянии от каждого из ядер. Если же потенциалы ионизации различны электронная плотность смещается в сторону атома с большим первым потенциалом ионизации[9]. Смещение электронной плотности приводит к асимметрии распределения электрических зарядов в молекуле, молекула становится полярной системой с определённым дипольным моментом.
Приближённые квантовохимические методы расчёта
Поскольку точное решение уравнения Шрёдингера для атомно-молекулярных систем, содержащих два и более электрона, невозможно, предложены приближённые методы расчёта электронной плотности. Все они возникли в 1930-х годах, проделали значительный путь развития и долгое время дополняли друг друга. Наиболее важные из них — теория валентных связей, теория молекулярных орбиталей, теория кристаллического поля, теория функционала плотности.
В рамках теории валентных связей разработана концепция резонанса (Л. Полинг) и родственная ей концепция мезомерии (К. Ингольд). Концепция резонанса рассмотрена на примере молекулярного иона водорода. Л. Полинг показал, что стабильность молекулярного иона водорода:
вызвана резонансом электрона, то есть движением его «взад и вперёд» между двумя ядрами с «резонансной частотой», равной по величине энергии резонанса (50 ккал/моль), делённой на константу Планка h. Для молекулярного иона в основном состоянии эта частота равна 7.1014сек−1[10].
Концепция резонанса дополняла постулаты классической теории химического строения и утверждала, что если для данного соединения классическая теория допускает построение нескольких приемлемых структурных формул, то действительному состоянию молекул этого соединения отвечает не какая-либо одна отдельная формула, а некоторое их сочетание (наложение, резонанс структур)[11].
Мезомерия является теорией электронного строения химических соединений, согласно которой истинное распределение электронной плотности в молекуле является промежуточным между распределениями, представленными несколькими классическими формулами[12].
Обычно рассматривают положительный и отрицательный мезомерные эффекты:
Винилхлорид: +М-эффект
Аллил-катион: −М-эффект
Кампания идеологического вмешательства в теорию химического строения
Кампания началась в 1949 году с публикации статьи В. М. Татевского и М. М. Шахпаронова «Об одной махистской теории в химии и её пропагандистах»[13]. В качестве главного объекта нападения была выбрана теория резонанса Л. Полинга. Было объявлено, что «представления о реальной молекуле как о чём-то среднем между двумя (и более) крайними абстрактными структурами являются буржуазными и поэтому направленными против всего самого „святого“». Были указаны и пропагандисты теории Я. К. Сыркин и М. Е. Дяткина — авторы книги «Химическая связь и строение молекул», в которой нашла отражение теория резонанса.
В воздухе очередной раз запахло инквизицией. В этой тревожной обстановке ведущие химики страны собрались на Всесоюзное совещание по проблемам химического строения (1951 г., Москва). Стенограмма этого совещания — один из наиболее позорных документов, когда-либо созданных коллективными усилиями ученых, хранится в химических библиотеках всего мира, и от этого срама бог весть когда ещё удастся отмыться… До крови не дошло — спасла оттепель, начавшаяся весной следующего года. Я. К. Сыркин и М. Е. Дяткина, подготовленные недавними друзьями и коллегами к выдаче в качестве первых козлов отпущения, уцелели; более того Я. К. Сыркин в дальнейшем стал академиком[14].
Лайнус Полинг удостоен в 1954 году Нобелевской премии по химии «за исследование природы химической связи и её применение для определения структуры сложных соединений».
Однако консенсус в теории химического строения не был достигнут. В. М. Татевский в курсе «Строение молекул» (1977 г.) отмечал:
… полностью выпадают и «висят в воздухе» представления уходящих в прошлое, но всё ещё фигурирующих в литературе так называемых «теории резонанса» и «теории мезомерии», которые не имеют основы ни в классической теории химического строения, ни в законных приложениях классической физики к вопросам строения молекул, ни в квантовой механике[15].
Лишь в 1991 году проведён принципиальный анализ кампании по борьбе с теорией резонанса и было показано, что эта кампания «нанесла серьёзный ущерб престижу советской науки»[16].
Атомы в молекулах
В классической теории химического строения понятие атома в молекуле является изначальным. Интуитивно ясно, что атом в молекуле меняется, меняются и его свойства в зависимости от окружения этого атома, прежде всего ближайшего. Основным показателем является расстояние между атомами в молекуле, определяющее как длину химической связи, так и прочность химической связи.
В квантовой теории понятие атома вторично. Так, по утверждению В. М. Татевского, молекула не состоит из атомов: «С современной точки зрения ясно, что при образовании молекулы сохраняются не атомы, а только ядра атомов и электроны»[17].
Было предпринято множество попыток сохранить понятие атома в молекуле, но, тем не менее, практически всегда они не удовлетворяли последующих исследователей по тем или иным причинам.
Одна из наиболее удачных попыток сохранения классической концепции атома в молекуле принадлежит Р. Бейдеру и его сотрудникам[18]. В рамках этой теории (QTAIM) электронная плотность «задаёт некоторое скалярное поле в трёхмерном пространстве, которое может быть охарактеризовано, например, его совокупностью экстремальных точек, линий и поверхностей, особых точек и т. п.»[19].
Таким образом, в квантовой теории атомов в молекулах Р. Бейдера оказывается возможным физическое обоснование ключевых понятий химии «атом», «молекула», «химическая связь» в терминах топологии функции электронной плотности в трёхмерном пространстве и описание химического строения молекул.
Электронная корреляция и конфигурация молекул
Электронная корреляция (взаимная обусловленность движения всех электронов атомной или молекулярной системы как целого определяется электростатическим отталкиванием электронов и статическим особенностями систем, в частности принципом Паули (фермиевская корреляция). Полный учёт электронной корреляции при расчёте энергии и определении электронной структуры системы достигается методом конфигурационного взаимодействия.
Простая и надёжная система правил для понимания и предсказания конфигурации молекул заложена в теории отталкивания электронных пар, наиболее важное правило которой достаточно эмпирично, хотя имеет квантовомеханическое обоснование, заключающееся в принципе Паули, а именно «электронные пары принимают такое расположение на валентной оболочке атома, при котором они максимально удалены друг от друга». При этом конфигурация молекулы будет определяться числом связывающих и неподелённых электронных пар у центрального атома:
- две связывающие электронные пары дают линейную конфигурацию молекулы;
- три — конфигурацию правильного треугольника;
- четыре — конфигурацию тетраэдра;
- пять — тригонально-бипирамидальную конфигурацию;
- шесть — конфигурацию октаэдра.
Наличие неподелённых электронных пар у центрального атома приводит к расширению типов конфигураций молекулы[20].
Строение соединений благородных газов
Молекула дифторида ксенона Молекула тетрафторида ксенона Молекула гексафторида ксенонаОткрытие Н. Бартлеттом в 1962 году первого соединения ксенона положило начало интенсивному развитию химии благородных газов, что привело к получению большого ряда соединений инертных газов и установлению их химического строения.
Оказалось, что все соединения благородных газов имеют обыкновенные ковалентные связи и общепринятые конфигурации молекул. Так, конфигурация молекулы дифторида ксенона линейна: связи ксенон—фтор являются аксиальными и взаимодействуют с тремя неподелёнными электронными парами, находящимися в экваториальных положениях.
В молекуле тетрафторида ксенона осуществляется конфигурация с плоским квадратным расположением лигандов. Неподелённые электронные пары ксенона максимально удалены друг от друга и расположены по разные стороны от этого квадрата.
Структура молекулы гексафторида ксенона представляет собой искажённый октаэдр. «Отклонение атомов фтора от вершин правильного октаэдра указывает на то, что неподелённая электронная пара занимает положение над центром одной из граней октаэдра, а атомы фтора, расположенные в углах этой грани, вынуждены раздвинуться»[20].
Открытие Бартлетта показало ошибочность популярного в то время правила октета,[как?] согласно которому при образовании молекул атомы удовлетворяют свою потребность в достижении 8-электронной валентной оболочки, подобной электронной конфигурации благородных газов за счёт попарного обобществления своих валентных электронов. Оказалось, что атомы благородных газов, имея полностью заполненную валентную оболочку, могут вступать в химические реакции и участвовать в химическом строении молекул.
Строение электронодефицитных соединений
Структурная проблема, связанная с электронодефицитными соединениями, довольно сложна. Фундаментальная трудность заключалась в том, что в молекулах нет достаточного числа валентных электронов для того, чтобы связать все атомы обычными двухэлектронными связями. Так, например, в молекуле диборана имеется двенадцать валентных электронов, все двенадцать нужны для образования ковалентных связей шести атомов водорода с бором, так что для связи атомов бора между собой электронов не остаётся. Сам Полинг допускал, что в диборане функционируют одноэлектронные связи, а молекула в основном состоянии резонирует между семью структурами льюисовского типа, а также между многочисленными структурами, содержащими одноэлектронные связи.
Однако заслуженное признание получили исследования природы химической связи в бороводородах, выполненные американским физикохимиком У. Липскомбом. В его интерпретации в диборане имеют место четыре двух- и две трёхцентровые связи.
Четыре концевые двухцентровые двухэлектронные связи HB лежат в одной плоскости. Два же центральных атома водорода расположены симметрично над этой плоскостью и под нею и объединены с атомами бора двумя трёхцентровыми связями.
В 1976 году Липскомб удостоен Нобелевской премии по химии с формулировкой «за исследование структуры боранов (боргидритов), проясняющих проблему химических связей».
Особенности строения сэндвичевых соединений
Молекула ферроценаДальнейшее развитие теории химического строения связано с открытием и установлением структуры ферроцена[21]. Оказалось, что при взаимодействии хлорида двухвалентного железа и циклопентадиена вместо ожидаемого соединения с двумя химическими связями железо-углерод образуется сэндвичеобразное соединение, в котором все 10 атомов углерода соединены с атомом железа.
Рентгеноструктурными исследованиями Э. Фишера установлено, что все атомы углерода в молекуле ферроцена структурно и химически эквивалентны. Атом металла взаимодействует не с одним или двумя конкретными атомами углерода, а со всеми атомами углерода двух циклопентадиенильных колец сразу. Атом металла как бы завис в пространстве между двумя циклами, представленных параллельными правильными пятиугольниками.
В настоящее время получены металлоцены — бициклопентадиенильные соединения переходных металлов для большинства d-элементов.
Сэндвичевые соединения образуются также с использованием в качестве органического фрагмента бензола или циклооктатетраена. Так, например, в ураноцене (см. рисунок) атом урана удерживает два восьмичленных кольца. Все 16 связей уран—углерод в ураноцене идентичны.
В 1973 году Э. Фишер и Дж. Уилкинсон удостоены Нобелевской премии по химии с формулировкой «За новаторскую, проделанную независимо друг от друга, работу в области металлорганических, так называемых сэндвичевых соединений».
Считается, что в сэндвичевых соединениях реализуется многоцентровая связь, тип химической связи, в которой в отличие от двухцентровой двухэлектронной связи, связывающие электронные пары распределены в пространстве трех или большего числа атомных центров молекулы, иона, радикала.[22]
Примечания
- ↑ 1 2 Совещание по теории химического строения в органической химии. Успехи физических наук.. — 1951, октябрь. — Т. XLV, вып. 2. — С. 277—392.
- ↑ Химический энциклопедический словарь / гл. ред. И. Л. Кнунянц. — Сов. энциклопедия, 1983. — С. 652. — 792 с.
- ↑ Butlerow A. Einiges über die chemische Structur der Körper. (Vorgetragen in der chemischen Section der 36. Versammlung deutscher Naturforscher und Aerzte zu Speyer am 19.Septbr.) (нем.) // Zeitschrift für Chemie und Pharmacie : magazin. — 1861. — Bd. 4. — S. 549—560.
- ↑ Бутлеров А. О химическом строении веществ // Учёные записки Казанского университета (отд. физ.-мат. и мед. наук). Вып.1, отд.1. — 1862. — С. 1—11.
- ↑ Бор Н. Избранные научные труды. — М.: «Наука», 1970. — Т. 1. — С. 147. — 584 с.
- ↑ 1 2 3 Коулсон К. Успехи физических наук (статья «Межатомные силы — от Максвелла до Шрёдингера»). — 1963, ноябрь. — Т. LXXXI, вып.3. — С. 545-556.
- ↑ Шусторович Е. М. Химическая связь. Сущность и проблемы.. — М.: «Наука», 1973. — С. 6. — 232 с.
- ↑ Потапов А. А. Ренессанс классического атома. — М.: «Наука», 2011. — С. 352. — 444 с.
- ↑ Ганкин В. Ю., Ганкин Ю. В. Как образуется химическая связь и протекают химические реакции. — М.: «Граница». — С. 65. — 320 с.
- ↑ Паулинг Л. Природа химической связи. — М.-Л.: Химическая литература, 1947. — С. 25—26. — 440 с.
- ↑ Новая иллюстрированная энциклопедия. Пр-Ро. Кн.15. — М.: Большая российская энциклопедия, 2002. — С. 169. — 255 с.
- ↑ Новая иллюстрированная энциклопедия. Ма-Мо. Кн.11. — М.: Большая российская энциклопедия, 2002. — С. 141. — 255 с.
- ↑ Вопросы философии. — 1949. — № 3. — С. 176—192.
- ↑ Охлобыстин О. Ю. Жизнь и смерть химических идей. Очерки по истории теоретической химии.. — М.: Наука, 1989. — С. 184. — 190 с.
- ↑ Татевский В. М. Строение молекул.. — М.: Химия, 1977. — С. 10. — 512 с.
- ↑ Сонин А. С. Печальный юбилей одной кампании. (рус.) // Вестник РАН. — 1991. — Т. 61, № 8. — С. 96—107. Архивировано 5 мая 2010 года.
- ↑ Татевский В. М. Квантовая механика и теория строения молекул. — М.: Издательство МГУ, 1965. — С. 17. — 162 с.
- ↑ Бейдер Р. Атомы в молекулах. Квантовая теория. — М.: Мир, 2001. — 532 с.
- ↑ Степанов Н. Ф. Квантовая механика и квантовая химия. — М.: Мир, 2001. — С. 487. — 519 с.
- ↑ 1 2 Р. Гиллеспи. Геометрия молекул. — М.: Мир, 1975. — 278 с.
- ↑ Несмеянов А.Н. Ферроцен и родственные соединения. Избранные труды 1969 — 1979. — М.: Советская энциклопедия, 1982.
- ↑ Химический Энциклопедический словарь. — М.: Советская энциклопедия, 1983. — 792 с.
ru-wiki.org
|
|
К шестидесятым годам прошлого столетия в органической химии накопился огромный фактический материал, который требовал объяснения. На фоне беспрерывного накопления экспериментальных фактов особенно остро проявлялась недостаточность теоретических представлений органической химии. Теория отставала от практики, от эксперимента. Такое отставание болезненно отражалось на ходе экспериментальных исследований в лабораториях; химики проводили свои исследования в значительной мере наугад, вслепую, зачастую не понимая природы синтезированных ими веществ и сути реакций, которые приводили к их образованию. Так, например, англичанин В. Перкин старш., синтезировавший в1856 г. краситель мовеин окислением нечистого анилина, совершенно не представлял себе механизма открытой им реакции; к тому же он вовсе не ставил перед собой задачу синтеза красителя, а пытался получить хинин. Органическая химия, по меткому выражению Вёлера, напоминала дремучий лес, полный чудесных вещей, огромную чащу без выхода, без конца. Насущные задачи органической химии требовали разрешения основного вопроса: являются ли молекулы беспорядочным нагромождением атомов, удерживаемых силами притяжения, или же молекулы представляют собой частицы с определенным строением, которое можно установить, исследуя свойства вещества. Теория типов Жерара, с теми или иными оговорками признававшаяся большинством химиков того времени, отказывалась на основании изучения химических свойств решать вопрос о строении молекул. А между тем в органической химии к этому времени уже накопились факты и обобщения, которые могли служить основой для решения этого вопроса. Так, например, теория радикалов дала органической химии чрезвычайно важное обобщение, заключавшееся в том, что при химических реакциях некоторые группы атомов в неизменном виде переходят из молекул исходных веществ в молекулы, образующиеся при этих реакциях. Теория типов, со своей стороны, в значительной мере способствовала изучению наиболее изменчивых частей молекул и причин этой изменчивости. Необычайно большое значение имело открытие валентности элементов. Исследуя состав металлоорганических соединений, Франк-ланд (1853) нашел, что каждый металл дает соединения со строго определенным числом радикалов; это число и представляет собой валентность данного металла. Ниже приведены простейшие металлоорганические соединения одно-, двух-, трех- ичетырехвалентных металлов: После открытия Франкланда стало ясно, что атомы могут соединяться в молекулы только в отношениях, определяемых валентностью атомов. В частности, было установлено, что углерод четырехвалентен (Кекуле, Кольбе). Открытие валентности непосредственно подводило кмысли, что молекулы имеют определенное строение. Однако вопросы о путях установления строения молекул, а также о зависимости свойств вещества от строения его молекул оставались открытыми. Новая эпоха в органической химии наступила с возникновением теории химического строения. Главная роль в создании. обосновании и подтверждении теории строения принадлежит знаменитому русскому ученому Александру Михайловичу Бутлерову, хотя кроме него элементы этой теории начали разрабатывать А. Купер (1831—1892) в Англии и А. Кекуле (1829— 1896) в Германии. В 1858 г. Купер опубликовал на трех языках (английском, французском и немецком) статью «О новой химической теории», где он отбрасывает теорию типов и высказывает точку зрения, согласно которой все особенности органических веществ могут быть объяснены, если учитывать только два свойства атомов: «избирательное сродство» (связь атомов) и «степень сродства» (валентность атомов). Купер писал: «С моей точки зрения этих двух свойств достаточно для объяснения всего того, что характерно для органической химии: именно это я докажу ниже… В молекуле, состоящей из трех, четырех, пяти и т. д. атомов углерода и эквивалентного количества водорода, кислорода и др., последние могут быть заменены другими элементами, в то время как углерод образует взаимно-связанный узел. Это означает, что один углерод связан с другим углеродом. Такое свойство придает углероду, так сказать, своеобразную физиономию и дает возможность понять непонятный до этого факт наслоения атомов углерода в органических соединениях». Придя таким образом к важному представлению о цепи углеродных атомов, Купер выражает далее свои взгляды в формулах, которые, по его замыслу, должны дать картину строения соединений. В качестве примера его формул, которые были первыми конституционными формулами, можно привести следующие: Из этих примеров видно, что Куперу удалось удивительно правильно передать конституцию этих соединений, а также некоторых более сложных и в то время мало исследованных (винная и виноградная кислоты). Однако все эти формулы были лишены опытного обоснования. Купер совершенно не ставил вопроса о возможности их экспериментальной проверки. Его формулы, как легко видеть, были основаны на формальной интерпретации понятий валентности и связи атомов, а отчасти даже на интуиции. Естественно, что при таком подходе невозможно избежать ошибок. Так, например, формулы глицерина, глицериновой кислоты и щавелевой кислоты, данные Купером, уже неверны: Таким образом, взгляды Купера, развитые им в его талантливой, интересной работе, не носят характера строгой теории. Другая попытка изображения органических соединений конституционными формулами была сделана в 1861 г. Лошмидтом. При построении своих формул Лошмидт рассматривал атомы как мельчайшие материальные частицы, подвергающиеся действию сил притяжения и отталкивания. Эти силы при сближении атомов уравновешиваются, и различные атомы удерживаются друг около друга в некотором равновесном положении. Сферы действия атомных сил Ло-шмидт условно обозначал кружками (например, атомы углерода и водорода — простыми кружками, кислорода—двойными, азота—тройными). Формулы Лошмидта имели следующий вид: Не пытаясь составить какое-либо представление о способе связи шести углеродных атомов в молекуле бензола, Лошмидт обозначал бензол символом В отличие от Купера, Лошмидт при выборе формул, кроме валентности («поллентности» по его выражению), иногда руководствовался и химическими свойствами. Однако в целом метод вывода формул Лошмидта был абстрактным, а зачастую просто необоснованным. Так, не опираясь на химические данные, Лошмидт пытался вывести формулы таких сложных веществ, как индиго, мочевая кислота и т. п. Естественно, что эти формулы оказались ошибочными. Несмотря на то, что многие из предложенных Лошмидтом формул органических соединений оказались удачными, работа его осталась почти не замеченной химиками того времени и не оказала сколько-нибудь существенного влияния на развитие теории органической химии. Большой вклад в создание структурной теории внес знаменитый немецкий химик Кекуле. Он установил четырехвалент-ность углерода, ввел тип метана, предложил известную формулу бензола и, что особенно важно, правильно сформулировал одну из основных задач органической химии того времени. В статье «О конституции и превращениях химических соединений и о химической природе углерода» в 1858 г. Кекуле писал: «Я считаю, что в настоящее время главной задачей химии не является обнаружение атомных групп, которые, вследствие некоторых своих свойств, могут быть рассматриваемы как радикалы, и причисление соединений к некоторым типам, которые едва ли при этом имеют иное значение, кроме как образца формулы. Напротив, я полагаю, что необходимо распространить размышление и на строение самих радикалов; из природы элементов должна быть выведена как природа радикалов, так и их соединений». Исходным пунктом для этого Кекуле считал «основность элемента» (валентность), а в отношении органических соединений прежде всего — природу углерода. Кекуле высказал также ряд других верных мыслей о связи атомов, выражая ее графическими формулами. Однако своим формулам Кекуле не придавал значения формул строения, он стремился выразить ими только реакционную способность. Так, он писал: «Рациональные формулы имеют своей целью дать определенное представление о химической природе соединения, следовательно, о его метаморфозах и отношениях, в которых оно находится к другим телам… При этом, естественно, необходимо иметь в виду, что рациональные формулы — это лишь формулы превращений, а не конституционные формулы, что они являются лишь средством выражения для метаморфоз тел и результатов сравнения различных веществ между собою; что они ни в коем случае не должны выражать конституцию, т. е. расположение атомов в соответствующем соединении». Установление истинного строения молекул Кекуле также считал задачей химии, но, по его мнению, это может быть достигнуто не путем изучения химических превращений, а только сразнительным исследованием физических свойств соединений. Таким образом, и в этом вопросе Кекуле стоял на позиции Же-рара. Как и другие сторонники теории типов, Кекуле изображал вещество несколькими типическими формулами. Так, например, чтобы передать известные тогда химические свойства уксусной кислоты, Кекуле предлагал изображать ее восемью формулами. Таким образом, хотя взгляды Кекуле и были близки к новым структурным воззрениям, хотя Кекуле и внес существенный вклад в развитие теории химического строения, он так и не смог полностью освободиться от представлений теории типов. А. М. Бутлеров выступил против положения теории типов о невозможности установления строения молекул химическим путем; он показал, что в молекуле имеется определенная последовательность химической связи атомов (химическое строение). Далее Бутлеров доказал, что строение молекулы можно установить, исследуя химические свойства вещества, и, наоборот, зная строение, можно предвидеть многие свойства соединения. Бутлеров не только обосновал это положение уже имевшимся фактическим материалом, но и предсказал на его основе возможность существования новых веществ, которые впоследствии были открыты им и другими химиками. Основная идея теории А. М. Бутлерова сформулирована им в 1861 г. в статье «О химическом строении веществ». Он писал: «Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определенным количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которого химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу». Основой теории Бутлерова является идея о порядке химического взаимодействия атомов в молекуле. Этот порядок химического взаимодействия не включает представления о механизме химической связи и физическом расположении атомов. Эта важная особенность теории химического строения позволяет всегда опираться на нее при построении физической модели молекулы. Установив понятие химического строения, А. М. Бутлеров дает новое определение природы вещества: «химическая натура сложной частицы определяется натурой элементарных составных частей, количеством их и химическим строением». Таким образом, А. М. Бутлеров первый установил, что каждая молекула имеет определенное химическое строение, что строение определяет свойства вещества и что, изучая химические превращения вещества, можно установить его строение. Взгляды А. М. Бутлерова на значение химических структурных формул вытекают из основных положений его теории. Бутлеров считал, что эти формулы должны быть не «типическими», «реакционными», а конституционными. В этом смысле для каждого вещества возможна лишь одна рациональная формула, на основании которой можно судить о химических свойствах. Что же касается способа написания структурных формул, то Бутлеров справедливо считал этот вопрос второстепенным: «Помня, что дело не в форме, а в сущности, в понятии, в идее,— и принимая во внимание, что формулами, обозначающими изомерию, логически-необходимо выражать настоящее частицы, т. е. некоторые химические отношения, в ней существующие,— не трудно притти к убеждению, что всякий способ писания может быть хорош, лишь бы только он с удобством выражал эти отношения. Весьма естественно даже употреблять разные способы, предпочитая тот, который является более выразительным для данного случая. Например, этан С2Н6 почти совершенно безразлично может быть изображен: …Однако же, при недостаточно-определенном понимании, иной способ писания может привести к недоумениям». С возникновением теории химического строения органическая химия вышла из лабиринта типических формул; были показаны пути к познанию внутреннего строения молекул; появилась теоретическая основа для понимания химических процессов, для предсказания новых путей синтеза органических соединений. С самого момента своего возникновения теория химического строения дала возможность химикам проводить экспериментальные исследования направленно, целеустремленно. Замечательным успехом теории химического строения явилось объяснение явления изомерии, открытого в первой четверти прошлого столетия. Как известно, в конце XVIII века был установлен закон постоянства состава, согласно которому каждое данное вещество имеет определенный, постоянный состав. В течение нескольких десятков лет этому закону придавалось и обратное значение, т. е. считалось,что данным определенным составом обладает лишь одно определенное вещество. Неправильность последнего положения была показана в результате исследования ряда органических веществ. В 1823 г. Либих, исследовав серебряную соль гремучей кислоты, установил, что ее состав (AgCNO) тождествен составу полученного в 1822 г. Вёлером и резко отличного от нее изоциановокислого серебра. Этот замечательный факт недолго оставался единичным; вскоре были обнаружены многие другие вещества, обладающие одинаковым составом, но разными свойствами. Открытое явление с 1830 г. начали называть изомерией (от греч. — составленный из одинаковых частей), а вещества с одинаковым составом — изомерами. Попытки объяснить изомерию теорией радикалов и теорией типов были односторонними (как и сами эти теории), а потому не давали удовлетворительных результатов. Фактически в течение почти четырех десятилетий явление изомерии не находило теоретического объяснения. Такое объяснение стало возможным только после создания теории химического строения, согласно которой природа вещест-ва определяется не только характером и числом атомов, входящих в состав молекулы, но и ее структурой, химическим строением. Отсюда с несомненной очевидностью вытекает возможность существования веществ, имеющих одинаковый состав и молекулярный вес и, тем не менее, совершенно различных вследствие неодинакового химического строения. Таким образом, различие в химическом строении явилось естественным и простым объяснением явления изомерии. Следует отметить, что А. М. Бутлеров открыл и впервые объяснил также явление динамической изомерии, заключающееся в том, что два или несколько изомеров в определенных условиях легко переходят друг в друга (явление это в настоящее время носит название таутомерия). Проблема изомерии в целом явилась серьезным испытанием для бутлеровской теории и была ею блестяще решена. Важной особенностью учения А. М. Бутлерова является то что он отнюдь не считал молекулу каким-то неподвижным образованием, в котором отдельные атомы связаны в мертвую, безжизненную конструкцию. По этому поводу он писал: «…в настоящее время мы смотрим на химическое соединение не как на что-либо мертвое, неподвижное; мы принимаем, напротив, что оно одарено постоянным движением, заключенным в его самых мельчайших частичках, частные взаимные отношения которых подлежат постоянным переменам, суммируясь при этом в некоторый постоянный средний результат. Мы можем иметь здесь и постоянные изменения в химических частицах, составляющих массу веществ, но все это сводится к известному среднему состоянию самой массы. Словом, вообще мы имеем всегда перед собою состояние известного подвижного равновесия. С этой динамической точки зрения на натуру химического соединения и на химические реакции, мы ясно объясняем такие явления, которые, с прежней точки зрения, были совсем непонятны. Стоит указать, например, на диссоциацию,— на то, как легко мы теперь объясняем обратные реакции и т. п.». Сформулированные А. М. Бутлеровым ясные, неопровержимые положения теории химического строения в короткий срок обеспечили ей всеобщее признание. Однако вместе с тем появилась тенденция замолчать заслуги А. М. Бутлерова и представить творцами теории строения только Кекуле и Купера. Уже несколько лет спустя после создания теории строения А. М. Бутлерову пришлось выступить в защиту своего приоритета, так как некоторые зарубежные химики, вначале не признававшие и даже не понимавшие его теории, впоследствии пытались приписать честь создания основных положений этой теории себе. Решающую роль А. М. Бутлерова в создании теории химического строения ярко подчеркнул в 1868 г. великий русский ученый Д. И. Менделеев, рекомендуя А. М. Бутлерова в Петербургский университет. Менделеев писал, что Бутлеров «…вновь стремится, путем изучения химических превращений, проникнуть в самую глубь связей, скрепляющих разнородные элементы в одно целое, придает каждому из них прирожденную способность вступать в известное число соединений, а различие свойств приписывает различному способу связи элементов. Никто не проводил этих мыслей столь последовательно, как он, хотя они и проглядывали ранее… Для проведения того же способа воззрения через зсе классы органических соединений Бутлеров издал в 1864 г. книгу: «Введение к полному изучению органической химии», в прошлом году переведенную на немецкий язык Бутлеров чтениями и увлекательностью идей образовал вокруг себя в Казани школу химиков, работающих в его направлении. Имена Марковникова, Мясникова, Попова, двух Зайцевых, Моргунова и некоторых других успели получить известность по многим открытиям, сделанным преимущественно благодаря самостоятельности бутлерозского направления. Могу лично засвидетельствовать, что такие ученые Франции и Германии, как Вюрц и Кольбе, считают Бутлерова одним из влиятельнейших в наше время двигателей теоретического направления химии». А. М. Бутлеров справедливо считал, что теория химического строения будет развиваться по мере накопления нового фактического материала. Он писал: «…не могу не заметить, что те заключения, к которым ведет принцип химического строения, оказываются в тысячах случаев согласными с фактами. Как во всякой теории, и здесь, конечно, есть недостатки, несовершенства, — встречаются факты, которые не отвечают строго понятию о химическом строении. Разумеется, следует желать в особенности размножения таких именно фактов; факты, не объясняемые существующими теориями, наиболее дороги для науки, от их разработки следует по преимуществу ожидать ее развития в ближайшем будущем»). Теория химического строения создавалась А. М. Бутлеровым в середине XIX века, в период, когда в России вырастали но-зые буржуазные общественно-экономические отношения и рост производительных сил привел к мощному развитию естествознания. В этот период И. М. Сеченов и затем И. П. Павлов создают материалистическое учение о высшей нервной деятельности человека и животных, К. А. Тимирязев и несколько позднее И. В. Мичурин закладывают начало нового этапа в развитии биологии, Д. И. Менделеев открывает важнейший закон природы — периодический закон, обобщивший все имевшиеся в то время знания о химических элементах. Н. И. Лобачевский открывает новую область математики. Теория химического строения создала возможность научной систематизации фактического материала органической химии, объяснила ее важнейшие закономерности и дала ключ к предсказанию новых фактов. Она явилась научной основой для создания современной органической химии. Предыдущая страница | Сдедующая страница СОДЕРЖАНИЕ |
www.xumuk.ru
Урок-лекция «Теория химического строения А.М. Бутлерова»
<Приложение 1. Слайд 1>
Задачи лекции:
- Образовательные:
- формировать понятия о сущности теории
химического строения органических веществ,
опираясь на знания учащихся об электронном
строении атомов элементов, их положении в
Периодической системе Д.И. Менделеева, о
степени окисления, природе химической связи и о
других главнейших теоретических положениях:
- последовательность расположения атомов углерода в цепи,
- взаимное влияние атомов в молекуле,
- зависимость свойств органических веществ от структуры молекул;
- сформировать представление о ходе развития теорий в органической химии;
- усвоить понятия: изомеры и изомерия;
- разъяснить смысл структурных формул орг.веществ и их преимуществ перед молекулярными;
- показать необходимость и предпосылки создания теории химического строения;
- продолжить формирование навыков составления конспекта.
- формировать понятия о сущности теории
химического строения органических веществ,
опираясь на знания учащихся об электронном
строении атомов элементов, их положении в
Периодической системе Д.И. Менделеева, о
степени окисления, природе химической связи и о
других главнейших теоретических положениях:
- Развивающие:
- развивать мыслительные приемы анализа, сравнения, обобщения;
- развивать абстрактное мышление;
- тренировать внимание учащихся при восприятии большого по объему материала;
- выробатывать умения анализировать информацию и выделять наиболее важный материал.
- Воспитательные:
- с целью патриотического и интернационального воспитания привести учащимся исторические сведения о жизни и деятельности ученых.
ХОД УРОКА
1. Организацонная часть
– Приветствие
– Подготовка учащихся к уроку
– Получение сведений об отсутствующих.
2. Изучение нового
План лекции: <Приложение 1. Слайд 2>
I. Доструктурные теории:
– витализм;
– теория радикалов;
– теория типов.
II. Краткая справка о состоянии химической науки к
60-м годам XIX столетия. Условия создания теории
химического строения веществ:
– необходимость создания теории;
– предпосылки теории химического строения.
III. Сущность теории химического строения
органических веществ А.М. Бутлерова. Понятие об
изомерии и изомерах.
IV. Значение теории химического строения
органических веществ А.М. Бутлерова и ее
развитие.
3. Задание на дом: конспект, п. 2.
4. Лекция
I. Знания об органических веществах
накапливались постепенно еще с глубокой
древности, но как самостоятельная наука
органическая химия возникла лишь в начале XIX
века. Оформление самостоятельности орг.химии
связано с именем шведского ученого Я. Берцелиуса
<Приложение 1. Слайд
3>. В 1808-1812 г.г. он издал свое большое руководство
по химии, в котором первоначально намеревался
рассмотреть наряду с минеральными также и
вещества животного и растительного
происхождения. Но часть учебника, посвященная
орг.веществам, появилась лишь в 1827 г.
Самое существенное различие между веществами
неорганическими и органическими Я. Берцелиус
видел в том, что первые могут быть получены в
лабораториях синтетическим путем, в то время как
вторые якобы образуются лишь в живых организмах
под действием некой «жизненной силы» –
химического синонима «души», «духа»,
«божественного происхождения» живых
организмов и составляющих их органических
веществ.
Теория, объяснявшая образование орг.соединений
вмешательством «жизненной силы», получила
название витализма. В течение
некоторого времени она пользовалась
популярностью. В лаборатории удавалось
синтезировать лишь самые простые
углеродсодержащие вещества, такие как
углекислый газ – СО2, карбид кальция – CaC2,
цианид калия – KCN.
Только в 1828 г. немецкий ученый Вёлер <Приложение
1. Слайд 4> сумел получить органическое
вещество мочевину из неорганической соли –
цианата аммония – NH4CNO.
NH4CNO ––t–> CO(NH2)2
В 1854 г. французский ученый Бертло <Приложение
1. Слайд 5>получил триглицерид. Это и
повлекло за собой необходимость изменения
определения органической химии.
Ученые пытались на основании состава и свойств
разгадать природу молекул органических веществ,
стремились создать систему, которая позволила бы
связать воедино разрозненные факты,
накопившиеся к началу XIX века.
Первая попытка создания теории, стремившейся
обобщить имевшиеся об орг.веществах данные,
связана с именем французского химика Ж.Дюма <Приложение 1. Слайд 6>.
Это была попытка рассмотреть с единой точки
зрения довольно большую группу орг.соединений,
которые сегодня мы называли бы производными
этилена. Орг.соединения оказывались
производными некоторого радикала C2H4
– этерина:
C2H4 * HCl – хлористый этил (солянокислый
этерин)
Заложенная в этой теории идея – подход к
орг.веществу как состоящему из 2-х частей – легла
в последствии в основу, более широкой теории
радикалов (Я. Берцелиус, Ю.Либих, Ф. Велер). Эта
теория основана на представлении о
«дуалистическом строении» веществ. Я.
Берцелиус писал: «каждое орг.вещество состоит
из 2-х составных частей, несущих противоположный
электрический заряд». Одной из этих составных
частей, а именно частью электроотрицательной,
Я.Берцелиус считал кислород, остальная же часть,
собственно органическая, должна была составлять
электроположительный радикал.
Основные положения теории радикалов:<Приложение 1. Слайд 7>
– в состав органических веществ входят
радикалы, несущие на себе положительный заряд;
– радикалы всегда постоянны, не подвергаются
изменениям, они без изменений переходят из одной
молекулы в другую;
– радикалы могут существовать в свободном
виде.
Постепенно в науке накапливались факты,
противоречащие теории радикалов. Так Ж.Дюма
провел замещение водорода хлором в
углеводородных радикалах. Ученым, приверженцам
теории радикалов, казалось невероятным, чтобы
хлор, заряженный отрицательно, играл в
соединениях роль водорода, заряженного
положительно. В 1834 г. Ж. Дюма получил задание
расследовать неприятное происшествие во время
бала во дворце французского короля: свечи при
горении выделяли удушливый дым. Ж.Дюма установил,
что воск, из которого делались свечи, фабрикант
для отбелки обрабатывал хлором. При этом хлор
входил в молекулу воска, заменяя часть
содержавшегося в ней водорода. Удушливые пары,
перепугавшие королевских гостей, оказались
хлороводородом (HCl). В дальнейшем Ж.Дюма
получил трихлоруксусную кислоту из уксусной.
Таким образом, электроположительный водород
заменялся крайне электроотрицательным
элементом хлором, а свойства соединения при этом
почти не менялись. Тогда Ж.Дюма сделал вывод, что
на место дуалистического подхода должен стать
подход к орг.соединению как единому целому.
Теория радикалов была постепенно отвергнута,
однако она оставила глубокий след в
органической химии: <Приложение
1. Слайд 8>
– понятие «радикал» прочно вошло в химию;
– верным оказалось утверждение о
возможности существования радикалов в свободном
виде, о переходе в огромном числе реакций
определенных групп атомов из одного
соединения в другое.
В 40-х г.г. XIXв. Было положено начало учению о гомологии, позволившему выяснить некоторые отношения между составом и свойствами соединений. Выявлены гомологические ряды, гомологическая разность, что позволило классифицировать органические вещества. Классификация орг.веществ на основе гомологии привела к возникновению теории типов (40-50-е годы XIX в., Ш. Жерар, А.Кекуле и др.) <Приложение 1. Слайд 9>
Сущность теории типов <Приложение 1. Слайд 10>
– в основу теории положена аналогия в реакциях между органическими и некоторыми неорганическими веществами, принятыми в качестве типов ( типы: водород, вода, аммиак, хлороводород и др.). Замещая в типе вещества атомы водорода на другие группы атомов, ученые предсказали различные производные. Например, замещение атома водорода в молекуле воды на радикал метил приводит к возникновению молекулы спирта. Замещение двух атомов водорода – к появлению молекулы простого эфира <Приложение 1. Слайд 11>
Ш. Жерар прямо говорил в связи с этим, что формула вещества – это только сокращенная запись его реакций.
Все орг. вещества считали производными простейших неорганических веществ – водорода , хлороводорода, воды, аммиака <Приложение 1. Слайд 12>
<Приложение 1. Слайд 13>
– молекулы органических веществ
представляют собой систему, состоящую из атомов,
порядок соединения которых неизвестен; на
свойства соединений влияет совокупность всех
атомов молекулы;
– невозможно познать строение вещества, так как
молекулы в процессе реакции изменяются.
Формула вещества отражает не строение, а
реакции, в которые данное вещество. Для
каждого вещества можно написать столько
рациональных формул, сколько различных видов
превращений может испытывать вещество.
Теория типов допускала множественность
«рациональных формул» для веществ в зависимости
от того какие реакции хотят этими формулами
выразить.
Теория типов сыграла большую роль в развитии органической химии <Приложение 1. Слайд 14>
– позволила предсказать и открыть ряд
веществ;
– оказала положительное влияние на развитие
учения о валентности;
– обратила внимание на изучение химических
превращений органических соединений, что
позволило глубже изучить свойства веществ, а
также свойства предсказываемых соединений;
– создала совершенную для того времени
систематизацию органических соединений.
Не следует забывать, что в действительности теории возникали и сменяли друг друга не последовательно, а существовали одновременно. Химики нередко плохо понимали друг друга. Ф.Вёлер в 1835 г. говорил, что «органическая химия в настоящее время может кого угодно свести с ума. Она представляется мне дремучим лесом полным чудесных вещей, огромной чащей без выхода, без конца, куда не осмеливаешься проникнуть…».
Ни одна из этих теорий не стала теорией органической химии в полном смысле слова. Главная причина несостоятельности этих представлений в их идеалистической сущности: внутреннее строение молекул считалось принципиально непознаваемым, а любые рассуждения о нем – шарлатанством.
Нужна была новая теория, которая бы стояла на материалистических позициях. Такой теорией явилась теория химического строения А.М. Бутлерова <Приложение 1. Слайды 15, 16>, которая создана в 1861 г. Все рациональное и ценное, что было в теориях радикалов и типов, было в дальнейшем ассимилировано теорией химического строения.
Необходимость появления теории диктовалась: <Приложение 1. Слайд 17>
– возросшими требованиями промышленности к
органической химии. Необходимо было
обеспечить текстильную промышленность
красителями. В целях развития пищевой
промышленности требовалось усовершенствовать
методы переработки сельскохозяйственных
продуктов.
В связи с этими задачами начали разрабатываться
новые методы синтеза органических веществ.
Однако у ученых возникли серьезные
затруднения по научному обоснованию этих
синтезов. Так, например, нельзя было объяснить
валентность углерода в соединениях с помощью
старой теории.
Углерод нам известен как элемент 4-х валентный
(Это было доказано экспериментально). Но здесь он
как будто только в метане CH4 сохраняет эту
валентность. В этане C2H6 если
следовать нашим представлениям, углерод д.б.
3-валентным, а в пропане C3H8 – дробную
валентность. (А мы знаем, что валентность должна
быть выражена только целыми числами).
Какова же валентность углерода в органических
соединениях?
Было непонятно, почему существуют вещества с одинаковым составом, но различными свойствами: С6H12O6 – молекулярная формула глюкозы, но такая же формула и фруктозы (сахаристого вещества – составной части мёда).
Доструктурные теории не могли объяснить многообразие органических веществ. (Почему углерод и водород – два элемента, – могут образовывать такое большое число различных соединений?).
Необходимо было систематизировать имеющиеся знания с единой точки зрения и разработать единую химическую символику.
Научно обоснованный ответ на эти вопросы дала теория химического строения органических соединений, созданная русским ученым А.М. Бутлеровым.
Основными предпосылками, подготовившими почву для возникновения теории химического строения были <Приложение 1. Слайд 18>
– учение о валентности. В 1853 г. Э. Франкланд ввел понятие о валентности, установил валентность для ряда металлов, исследуя металлоорганические соединения. Постепенно понятие валентности было распространено на многие элементы.
В 1858 г. А. Кекуле предложил считать атом углерода четырехвалентным.
Важным открытием для органической химии явилась гипотеза о способности атомов углерода к образованию цепей (А. Кекуле, А. Купер).
Одной из предпосылок была выработка правильного представления об атомах и молекулах. До 2-й половины 50-х г.г. XIXв. Не было общепризнанных критериев для определения понятий: «атом», «молекула», «атомная масса», «молекулярная масса». Только на международном конгрессе химиков в Карлсруэ (1860 г.) были четко определены эти понятия, что предопределило развитие теории валентности, возникновение теории химического строения.
Основные положения теории химического строения А.М. Бутлерова (1861 г.)
А.М. Бутлеров сформулировал важнейшие идеи теории строения органических соединений в виде основных положений, которые можно разделить на 4 группы.<Приложение 1. Слайд 19>
1. Все атомы, образующие молекулы органических веществ, связаны в определенной последовательности согласно их валентности (т.е. молекула имеет строение).
<Приложение 1. Слайды 19, 20>
В соответствии с этими представлениями валентность элементов условно изображают черточками, например, в метане CH4. <Приложение 1. Слайд 20>>
Такое схематичное изображение строения молекул называют формулами строения и структурными формулами. Основываясь на положениях о 4-х валентности углерода и способности его атомов образовывать цепи и циклы, структурные формулы орг.веществ можно изобразить так: <Приложение 1. Слайд 20>
urok.1sept.ru