Отрицание в немецком языке выражается при помощи Nein (Нет), nicht (не) и kein (никакой):
Haben Sie eine Kamera? Fotografieren Sie? – У Вас есть фотоаппарат? Вы фотографируете?
Ich habe eine Kamera. – У меня есть фотоаппарат.
Nein, ich habe keine Kamera. – Нет, у меня нет фотоаппарата. (Дословно: у меня никакой фотоаппарат.)
Как видите, при отрицании eine заменяется на keine (к eine просто приставляется буква k).
Если же артикль определенный (или если вместо него стоит притяжательное местоимение), то употребляется nicht, которое стремится встать на конец предложения:
Ich habe die Kamera nicht. – У меня нет этого фотоаппарата.
С глаголами, конечно, всегда nicht (никакой здесь не употребишь):
Nein, ich fotografiere nicht. – Нет, я не фотографирую.
Если вы хотите подчеркнуть, что у вас не этот фотоаппарат, а другой, то nicht ставится перед словом, которое вы подчеркиваете. Логическое ударение падает при этом на определенный артикль:
Ich habe nicht die Kamera (sondern eine andere). – У меня не этот фотоаппарат (а другой).
Слова, указывающие на образ действия (как?) или качество (каков?), т. е. прилагательные, не пропускают nicht дальше себя:
Die Kamera ist nicht gut. – Этот фотоаппарат не хорош.
Das Auto fährt nicht schnell genug. – Машина едет недостаточно быстро.
А также:
Sie ist nicht meine Frau. – Она не моя жена. (Здесь тоже качество (в качестве моей жены), но выражено оно существительным.)
Слова, указывающие на место (где? куда? откуда?), а также предлог + местоимение также мешают nicht встать на конец предложения:
Ich gehe nicht auf die Straße. – Я не пойду на улицу.
Sie wartet nicht auf mich. – Она не ждет меня (дословно: на меня).
Иногда слова могут употребляться вообще без артикля, хотя они его, конечно, имеют:
Trinken Sie Bier? – Вы пьете пиво?
Sprechen Sie Russisch? – Вы говорите по-русски?
Hast du Hunger? – Ты хочешь есть (дословно: имеешь голод)?
Отрицательный ответ будет таким, как если бы в вопросе был неопределенный артикль:
Nein, ich trinke kein Bier. Ich spreche kein Russisch. Ich habe keinen Hunger.
Ein и nicht могут сосуществовать лишь в том случае, если вы хотите выделить существительное и ставите его для этого в начало предложения:
Eine Kamera brauche ich nicht. – Фотоаппарат мне не нужен.
Bier trinke ich nicht. – Пиво я не пью.
www.de-online.ru
Отрицание в немецком: kein und nicht
Отрицание в немецком: kein und nicht
Давай-ка разберем в этой статье отрицание в немецком: kein и nicht, узнаем, когда и что нужно употреблять!
Начнем с Negationsartikel (отрицательный артикль) или как его еще называют отрицательного местоимения «kein»!
Когда мы употребляем kein?
«kein» используем в повествовательных предложениях в сочетании с существительными, которые имеют неопределенный артикль ein либо с существительными без артикля (с нулевым артиклем). Поэтому вместо ein/eine при отрицании ставим kein/keine перед нашим существителным.
Beispiele: Ist das ein Buch? — Nein, das ist kein Buch. Willst du eine Birne? — Nein, ich will keine (Birne можно опустить). Gloria hat keinen Fehler gemacht. Wir haben kein Tor geschossen. Wir haben keine Zeit. Ich habe keine Freunde. Lisa hat keine Angst. Sigmund hat keinen Hunger
Если перед существительным идет прилагательное, то схема следующая: kein + прилагательное + существительное Естественно, не забываем про склонение kein и прилагательных!
Beispiele:
Wir finden keine guten Bücher hier. Ich habe keine gute Idee Sie hat keine tolle Figur Meine Freundin hat keine schönen Klamotten
Нужно знать, что kein склоняется также, как и артикли:в ед. числе склонение будет таким же, как и у неопред-го артикля. А во множ. числе «kein» будет склоняться, как определенный артикль.
А теперь перейдем к отрицательной частичке «nicht».
Её употребление: Если наше существительное имеет определенный артикль, то тут уже делаем отрицание с частицей nicht. Beispiele:
Ist das dein Heft? — Nein, das ist nicht mein Heft. Ist sie die Lehrerin? — Nein, sie ist nicht die Lehrerin. Sie ist nicht meine Freundin!
Также, при помощи отрицательной частички «nicht» могут отрицаться другие члены предложения в немецком такие, как глаголы, прилагательные и др.
А куда в предложении ставить «nicht»?
Место «nicht» зависит в предложении от того, ЧТО вы хотите отрицать. Если мы отрицаем простое сказуемое или всю идею предложения, то эта частичка уходит в конец предложения: Beispiele:
Ich singe nicht. Leo kommt heute nicht. Ich besuche meinen Freund nicht. Ich helfe meiner Mutter am Wochenende nicht Verstehst du das? — Nein, ich verstehe das nicht. Angela tanzt nicht.
Если отрицается сложное сказуемое (состоящее из двух и более глаголов), то «nicht» ставится перед 2-ой частью глагола: Beispiele:
Sie hat dieses Buch noch nicht gelesen. Samantha wird diese Regeln nicht wiederholen.
Если отрицание относится к любому другому члену немецкого предложения, то частичку «nicht» ставим непосредственно перед членом предложения, который мы отрицаем:
если отрицается прилагательное, то «nicht» ставим сразу перед прилагательным:
Beispiele:
Laura ist nicht alt. Mona kocht nicht gut. Eduard ist nicht sportlich. Antonio ist nicht groß. Linda ist nicht dick. Wir laufen nicht schnell.
если отрицаем слова, указывающие на место (где? откуда и куда?):
Beispiele:
Ich fahre heute mit meiner Freundin nicht nach Dortmund (, sondern nach Stuttgart). Morgen bleiben wir nicht zu Hause. Er kommt nicht aus der Türkei.
если есть предлог + местоимение
Beispiele:
Sie wartet nicht auf mich. Wir gehen nicht auf die Straße. – Мы не пойдем на улицу.
nicht ставится перед именами собственными, если мы хотим их отрицать:
Beispiele:
Er ist nicht Herr Steinbeck. Ich bin nicht Frau Ecker! Ich bin Frau Becker!
Nicht и ein могут использоваться в одном и том же предложении лишь в двух случаях: -если говорящий хочет выделить существительное в предложении и ставит его для этого в начало предложения. При этом nicht будет стоять в самом конце: Eine Kamera braucht sie nicht. – Фотокамера ей не нужна. — если под Ein подразумевается обычное число 1. Ich habe nicht einen Euro in der Tasche. (= Ich habe keinen Euro in der Tasche.)
А если в кратце, то kein употребляется перед существительными, у которых нет артикля либо есть артикль ein/eine. А nicht употребляется с другими членами предложения такими, как прилагательные, глаголы.. (также и с сущ., у которых есть определенный артикль der/die/das либо если перед существительным стоит притяжательное местоимение mein/dein…). Ich esse kein Fleisch, weil ich Fleisch nicht mag. — Я не ем мясо, потому что не люблю его.
П.с. не забудь поделиться данной статейкой с другими хорошими людьми, нажав ниже соц.кнопочки=)) Благодарю тебя мой милый друг =))) Прекраснейшего тебе настроения!
Еще интересно:
Всё, что надо знать про Start Deutsch A1 (старт дойч а1)!
Длинные немецкие слова
Немецкие вспомогательные глаголы: Hilfsverben
Немецкий! Тема: Музыка, музыкальные инструменты и профессии на немецком языке. Musik und Musikinstrumente
Список немецких глаголов на уровень А1: неправильные глаголы на А1 + их спряжение в 3-х временах
Открытка из Германии На Удачу и С Любовью….=)
Тема Еда на немецком!
Немецкие глаголы с предлогами и падежами: управление немецких глаголов
Когда используем das Präteritum и das Perfekt? Какие глаголы употребляются только в Präteritum?
Интересно почитать
Вконтакте
Facebook
Twitter
Google+
Pinterest
Одноклассники
Мой мир
lifeistgut.com
Отрицание в немецком языке. Negation
Отрицание в немецком языке можно выразить с помощью отрицательных слов nicht, kein, weder … noch, nichts, niemand и так далее.
Ist das dein Fahrrad? — Nein. Ist das dein Auto? — Ja.
Ist das dein Fahrrad? — Nein, es ist nicht meins. Mein Fahrrad steht da drüben. Ist das dein Auto? — Ja, das ist mein Auto.
Ist das nicht dein Fahrrad? — Nein. Ist das nicht dein Auto? — Doch. (Das ist mein Auto)
Отрицание с помощью nicht. Место nicht в предложении
Nicht может отрицать всё предложение, глагол или существительное с определенным артиклем.
Если в предложении один глагол, и мы его отрицаем, то nicht стоит в самом конце предложения перед точкой.
Arbeitest du? – Nein, ich arbeite nicht. Kochst du das Mittagessen? – Nein, ich koche das Mittagessen nicht. Kommst du mit uns ins Kino heute Abend? – Nein, ich komme mit euch in Kino heute Abend nicht.
Если в предложении 2 глагола (глаголы с отделяемыми приставками, предложения с модальными глаголами, инфинитив, прошедшее время), то nicht стоит на предпоследнем месте.
Macht sie die Tür zu? – Nein, sie macht die Tür nicht zu. Hast du heute die Zeitung gelesen? – Nein, die habe ich heute noch nicht gelesen. Muss ich alle Vokabeln lesen? – Nein, du musst alle Vokabeln nicht lesen, du musst sie lernen.
Если отрицаем предлог, то nicht стоит перед предлогом.
Fährst du mit dem Zug nach Lübeck? – Nein, ich fahre nicht mit dem Zug nach Lübeck, ich fahre mit dem Auto. Geht er morgens ins Schwimmbad? – Nein, er geht nicht ins Schwimmbad, er joggt im Park. Kommen Sie aus Frankreich? – Nein, ich komme nicht aus Frankreich.
Если предлог стоит на 1 месте, то nicht стоит в самом конце предложения.
Nicht не может стоять в начале предложения!
Fährst du mit diesem Zug nach Lübeck? – Nein, mit diesem fahre ich nicht. Geht er morgens ins Schwimmbad? – Nein, ins Schwimmbad geht er nicht. Kommen Sie aus Frankreich? – Nein, aus Frankreich komme ich nicht.
Nicht стоит перед отрицаемыми словами (сегодня, много, просто так, охотно и т.д.).
Liest du viel? – Nein, ich lese nicht viel. Trinkst du Mineralwasser? – Nein, ich trinke Mineralwasser nicht gern. Ich mache diese Aufgabe nicht heute.
Отрицание с помощью nicht
Часто нужно отрицать не все предложение, а только определенную часть или одно слово. В этом случае nicht будет стоять перед тем, что отрицаем. Интонацией сильно выделяем отрицание nicht и то, что отрицаем. В некоторых случаях допустимо nicht в начале предложения. Если отрицаем какое-то слово или часть предложения, необходимо ввести альтернативу отрицанию (не сегодня, а завтра; не я, а он; не включить, а выключить и т.д.). Для этого используется оборот nicht… , sondern.
Nicht Sonja hat das Glas gebrochen, sondern Christine. Du liest dieses Buch jetzt, nicht morgen. Nicht am Freitag, sondern am Samstag beginnt der Wettbewerb. Er konnte nicht ein Stück, sondern gleich eine ganze Torte essen. Wir gratulieren nicht nur dir, sondern deiner ganzen Familie. Bitte, schalte das Licht in dem Zimmer nicht aus, sondern ein.
Nicht может отрицать прилагательное, причастие или группу прилагательных. В этом случае nicht будет стоять перед прилагательным.
Mein Freund trägt oft dieses nicht gebügelte Hemd. Die nicht lange dauernde Vorlesung hat das Interesse der Studenten geweckt. Du hast mir ein noch nicht gelesenes Buch gegeben.
Отрицание с помощью kein
Существительное с определенным артиклем отрицается с помощью nicht.
Существительное с неопределенным артиклем отрицается с помощью kein-.
Существительное без артикля отрицается с помощью kein-.
Отрицательный артикль kein- склоняется точно так же, как и неопределенный артикль.
Во множественном числе нет неопределенного артикля, есть только отрицательный артикль keine.
Kasus
Maskulinum
Femininum
Neutrum
Plural
Nominativ
kein
keine
kein
keine
Akkusativ
keinen
keine
kein
keine
Dativ
keinem
keiner
keinem
keinen
Genitiv
keines
keiner
keines
keiner
Ist das ein Buch? – Nein, das ist kein Buch, sondern ein Heft. Ist das ein Radiergummi? – Nein, das ist kein Radiergummi, sondern ein Spitzer. Sind das _ Schüler? – Nein, das sind keine Schüler, sondern _ Studenten. (Plural !) Hat er eine Freundin? – Nein, er hat keine Freundin, er ist Single.
Если перед существительным стоит числительное eins, то оно склоняется как неопределенный артикль. Числительное eins отрицается с помощью nicht.
Ich habe von meinen Eltern nicht ein Geschenk, sondern zwei. Helga hat nicht einen Computer zu Hause, sondern drei. Meine Mutter hat nicht eine Bananentorte gebacken, sondern fünf.
Отрицательные слова
положительно
отрицательно
Beispiele
Особа
jemand – кто-то
niemand – никто
Hast du da jemanden gesehen? — Nein, da habe ich niemanden gesehen.
Предмет
etwas, alles – что-то, все
nichts – ничего
Bestellst du etwas für sich? — Nein, ich bestelle nichts.
Время
jemals – когда-нибудь, oft – часто, immer – всегда, manchmal – иногда
nie, niemals – никогда
Wart ihr schon jemals in Österreich? — Nein, dort waren wir noch nie. In Österreich war ich niemals.
Место
irgendwo – где-то, überall – везде
nirgendwo, nirgends – нигде
Irgendwo in dem Flur liegt mein Regenschirm. Ich kann deine Brille nirgends finden.
Направление
irgendwohin – куда-то
nirgendwohin – никуда
Ich überlege mir, ob wir irgendwohin im Sommer in den Urlaub fahren. Mein Auto ist leider kaputt, ich kann jetzt nirgendwohin fahren.
Конструкции с отрицательным значением
«…ни …, ни … » («weder … noch»)
Tim kann nicht Deutsch sprechen. Er kann auch nicht Englisch sprechen. Tim kann weder Deutsch noch Englisch sprechen. – Тим не умеет говорить ни по-немецки, ни по-английски.
Meine kleine Schwester kann noch nicht lesen. Sie kann auch nicht schreiben. Meine kleine Schwester kann weder lesen noch schreiben. – Моя маленькая сестра не может ни читать, ни писать.
не сделав что-то (ohne … zu)
Paul will reisen. Er will nicht viel Geld ausgeben. Paul will reisen, ohne viel Geld auszugeben. – Пауль хочет путешествовать, не тратя много денег.
Sie geht weg. Sie verabschiedet sich nicht. Sie geht weg, ohne sich zu verabschieden. – Она уходит, не прощаясь.
Предлоги с отрицательным значением
без + падеж Аккузатив (ohne + Akkusativ)
Wir beginnen die Feier. Wir warten auf dich nicht. Wir beginnen die Feier ohne dich.
Der junge Mann fährt im Zug. Er hat keine Fahrkarte. Der junge Mann fährt im Zug ohne Fahrkarte.
кроме + падеж Датив (außer + Dativ)
Die ganze Touristengruppe ist pünktlich zum Bus gekommen, nur Herr Berger nicht. Die ganze Touristengruppe außer Herrn Berger ist pünktlich zum Bus gekommen.
Meine Freunde haben schon alles in dieser Stadt gesehen, nur das Rathaus nicht. Meine Freunde haben alles in dieser Stadt außer dem Rathaus gesehen.
Префиксы и суффиксы для отрицания
Префиксы стоят перед корнем и придают слову значение «не»:
apolitisch, asozial, atypisch Das war atypisch für ihn, kein Bier am Freitagabend zu trinken.
desillusioniert, desinfiziert, desinteressiert, desorganisiert, desorientiert Die Hotelzimmer sind desinfiziert und aufgeräumt.
indiskutabel, indiskret, inkompetent, instabil, intolerant Sein Zustand ist jetzt instabil. / Solches Verhalten ist intolerant.
irrational, irregulär, irreal, irrelevant, irreligiös, irreparabel Das Bild scheint irreal zu sein.
Entschuldigung, ich habe das unbewusst gemacht. Warum benimmst du dich so unfreundlich? Dieses Gerät ist für die regelmäßige Verwendung ungeeignet. Das ist sehr leicht, die Aufgabe ist unkompliziert.
Суффиксы стоят после корня и придают слову значение «без» или «не»:
Es macht keinen Sinn, ihm solche Witze zu erzählen, er ist total humorlos. Mein Freund wandert viel, er ist ein anspruchsloser Tourist, er kann im Zelt im Schlafsack schlafen. Weiter diese Geschichte zu erzählen war schon sinnlos. Sprachlos stand sie vor mir und konnte nicht verstehen, was passierte.
speakasap.com
Отрицание в немецком языке: для уровня А1 надо знать nicht и kein
Я из Москвы, но старшие классы школы оканчивала в Словакии. Немецкий учила с 7 класса, но он всегда казался довольно неприступным языком и зачастую вызывал панику. Но мне необходимо было получить сертификат уровня C1 для поступления в университет в Вене (Universität Wien). Поняла, что школьной программы мне не хватит, поэтому обратилась к Google, и наткнулась на сайт Екатерины Алексеевны.
Сам сайт очень грамотно оформлен, приложены сертификаты знания языка, рассчитаны чистые часы для освоения того или иного уровня. Это как-то сразу мотивировало и все показалось не таким уж безнадежным.
Среди множества репетиторов, которых я нашла, Екатерина Алексеевна показалась наиболее компетентным, располагающим к себе и знающим своё дело педагогом. Недолго думая, я обратилась к ней за помощью, и это было моим лучшим решением во всей этой неравной борьбе с немецким языком. Очень благодарна ей за понимание и готовность помочь. Все уроки были крайне интенсивны и продуктивны, курс был грамотно структурирован и организован, я впервые столкнулась с тем, что преподаватель настолько посвящён своему ученику.
Платформа для домашних заданий была очень удобна в использовании, и я до сих пор ее использую в случае чего, так как доступ остаётся открытым даже после окончания курса. Материал, который Екатерина Алексеевна мне предоставила был действительно очень полезен и разнообразен, все было крайне полезно для освоения немецкого.
Мой курс длился 3 месяца. Для начала, мне надо было подтянуть В2, так как он был в довольно плачевном и хаотичном состоянии. Первые пару-тройку занятий уже смогли внести ясность в мои школьные познания, и все начало обретать смысл. Освоив В2, мы начали подготовку к экзамену Goethe Zertifikat C1. Разница в уровнях была довольно ощутима, но тем не менее Екатерина всегда была готова все доступно объяснить.
Первый раз сдавала экзамен в Москве в институте Гёте, но мне не хватило одного балла за письменную часть, так что меня ожидала попытка номер два. Через месяц проводился экзамен в Саратове, в лингвистическом центре «Лингва-Саратов». И на этот раз я уже постигла С1, набрала 71 бал. За письменную часть 48 баллов и 23 балла за устную. Это не верх совершенства, ещё есть к чему стремиться. Мой путь к немецкому был тернист, но Екатерина мне очень помогла, безмерно ей благодарна.
В ВУЗ я успешно поступила, сейчас на первом семестре курса Japanologie.
Всем, у кого такие же тяжелые отношения с немецким, какие были у меня, рекомендую обратиться к Екатерине Алексеевне: очень тёплый и добрый человек, и первоклассный педагог.
a1c2deutschonline.com
Отрицание в немецком языке — Немецкий язык онлайн
В немецком языке есть несколько основных способов выразить отрицание.
Видео на данную тему:
Пожалуй, тему над открыть, сказав вам, что в немецком языке, в отличие от русского, невозможно двойное отрицание. Возможно, это правило не удивит тех, кто уже более или менее знаком с темой отрицания в немецком языке.
Немецкий язык не позволяет таких емких высказываний, как русское предложение: «Никому ничего не скажу».
В немецком языке приходится ограничиться одним отрицанием:
Ich werde es niemandem sagen. Я не скажу это никому.
Вам следует в первую очередь запомнить разницу между словами nein и nicht:
nein – означает всегда «нет», а
nicht – «не».
При письме надо не забывать ставить запятую после слова „Nein“
Ist der Termin am Dienstag? Nein, der Termin ist erst am Donnerstag!
Помимо этих отрицаний есть также еще одно отрицание, несвойственное для русского языка, отрицание существительных «kein», что означает «никакой».
Сейчас мы разберем на примерах в чем же отличие отрицаний «kein» и «nicht».
Kein отрицает только существительные с определенным артиклем или нулевым артиклем, что это такое, вы уже узнали из наших прошлых выпусков. Kein изменяется по родам и падежам:
Ich habe einen Hund. — У меня есть собака.
Nein, ich habe keinen Hund. — Нет, у меня нет собаки. (Дословно: у меня нет никакой собаки.)
Ich lese ein Buch. – Я читаю книгу.
Nein, ich lese kein Buch. (Дословно: я читаю никакую книгу.)
Ich lese eine Zeitung. – Я читаю газету.
Nein, ich lese keine Zeitung. (Дословно: я читаю никакую газету.)
Sie hat Zeit. – У нее есть время.
Sie hat keine Zeit. – У нее нет времени. (Дословно: Она имеет никакое время.)
Sprechen Sie Spanisch? – Вы говорите по-испански?
Ich spreche kein Spanisch. – Я не говорю по-испански.
Es gibt ein Problem. — Есть проблема.
Es gibt kein Problem. — Нет никакой проблемы.
Ich habe Hunger. – Я голоден/голодна.
Ich habe keinen Hunger. — Я не голоден/голодна.
Ich habe kein Wort verstanden. Я не понял/а ни одного слова.
Вы, вероятно, уже заметили, что отрицание kein стоит перед существительным и напоминает неопределенный артикль ein. Все верно, данное отрицание kein склоняется также, как и неопределенный артикль ein. Часто его еще называют отрицательным артиклем.
Здесь главное правило такое: отрицание „kein” употребляется всегда только с существительными, а также склоняется, как неопределенный артикль «ein».
Давате перейдем к следующему отрицанию: nicht.
Отрицание nicht употребляется тогда, когда отрицается не существительное, а другая часть речи, например, сказуемое, выраженное глаголом, дополнение, местоимение и прочее.
Ich wohne nicht in Dresden und ich heiße nicht Andrea.
Как правило, отрицание nicht стоит перед отрицаемым словом или словосочетанием:
Ich wohne nicht in Dresden.
Ich kaufe das Buch nicht für mich.
Также, отрицая все предложение, nicht может стоять в конце:
Das interessiert mich nicht. — Это меня не интересует.
Am Montag kann ich nicht. – В понедельник я не могу.
Mir gefällt der Film nicht. – Мне не нравится (этот) фильм.
Ich arbeite heute nicht. – Сегодня я не работаю.
Er kommt wahrscheinlich nicht. – Он вероятно не придет.
В случае, если в предложении есть 2 глагола, то nichtстоит перед вторым глаголом:
Sie ist gestern nicht gekommen. – Она не пришла вчера.
Leider kann ich dich nicht anrufen. – К сожалению, я не могу тебе позвонить.
Ich werde es nicht machen. — Я не буду этого делать.
Если в предложении с отрицанием nicht употребляется также глагол с отделяемой приставкой, то отрицание находится перед отделяемой приставкой, например:
Wir fahren heute nicht weg. – Мы сегодня не уезжаем.
Das nehme ich nicht mit. – Это я не возьму с собой.
Если встречаются, например, прилагательные или наречия, то nicht стоит тоже перед ними:
Das Essen ist nicht gut. – Еда невкусная.
Das finde ich nicht schlecht. – По-моему, это неплохо.
Есть еще одно применение отрицания nicht: конструкция «nicht…, sondern…» — не…, а…
Ich wohne nicht in Dresden, sondern in Köln. – Я живу не в Дрездене, а в Кёльне.
Das ist nicht unsere Wohnung, sondern die Wohnung von unseren Nachbarn. – Это не наша квартира, а квартира наших соседей.
Ich kaufe das Buch nicht für mich, sondern für meine Freundin.- Я покупаю книгу не себе (для себя), а моей (для моей) подруги.
В этой теме есть еще одна особенность: Если с существительным употребляется определенный артикль (или если вместо него стоит притяжательное местоимение (мой, моя, моё, мои и т.д.)), то вместо отрицания kein употребляется nicht, которое, как правило, стоит в конце предложения:
Das ist nicht unsere Wohnung. (нельзя сказать keine unsere – это является неверным) – Это не наша квартира.
Das Buch lese ich nicht . — (Эту) книгу я не читаю.
Также вы можете подчеркнуть информацию о том, что вы не читаете эту книгу, а испытываете интерес в чтении другой книги:
Ich lese nicht das Buch, sondern ein anderes.
Ich trinke Bier. — Я пью пиво.
Ich trinke kein Bier. – Я не пью пиво.
Bier trinke ich nicht. — Пиво я не пью.
Помимо этих отрицаний конечно же есть и другие:
immer (всегда) / irgendwann (когда-нибудь) — nie (никогда)
Die Jacke ist irgendwo (Куртка (находится) где-то). – Die Jacke ist nirgendwo (Куртки нигде нет).
Sie will irgendwohin mit dem Auto fahren (Я хочу куда-то поехать на машине). – Sie will nirgendwohin mit dem Auto fahren (Я не хочу никуда ехать на машине).
Er fährt irgendwoher (Он куда-то едет). — Er fährt nirgendwoher (Он никуда не едет).
Светлана Кижикова учитель курсов немецкого языка от Start DeutschИсточник: http://startdeutsch.ru/grammatika/artikl/955-artikli-stran-v-nemetskom
startdeutsch.ru
Особые правила положения nicht в предложении
28.03.2014 ПЯТНИЦА 13:29
ГРАММАТИКА
В этой статьи мы познакомимся с особыми правилами положения nicht в предложении – Besondere Regeln bei der Verwendung von „nicht“ im Satz.
1. При частичном отрицании nicht стоит:
перед отрицаемым словом:
Er fährt nicht mit dem Auto, sondern mit dem Bus.
Он поедет не на машине, а на автобусе.
перед словом, обозначающим качество/свойство:
Der Student arbeitet nicht fleißig.
Студент работает не прилежно.
в конце предложения, то есть не перед отрицаемым словом, если оно сильно выделяется за счёт интонации:
Gestern ist er nicht gekommen.
Вчера он не пришёл. — частичное отрицание.
Но: Gestern ist er nicht gekommen.
Вчера он не пришёл. — полное отрицание.
2.При полном отрицании nicht стоит:
в конце предложения, если сказуемое не имеет второй, неспрягаемой, отделяемой части:
Er liest die Zeitschrift nicht.
Он не читает журнал.
перед второй, неспрягаемой частью сказуемого:
Er wird die Zeitschrift nicht lesen.
Он не будет читать журнал.
Er liest die Zeitschrift nicht vor.
Он не читает вслух журнал.
в обязательном порядке перед существительным в Винительном падеже, если оно с глаголом составляет единое целое:
Er spielt nicht Klavier.
Он не играет на пианино.
Er nahm nicht Abschied.
Он не простился.
чаще перед дополнениями с предлогами:
Er denkt nicht daran.
Он не думает об этом.
Sie erinnert sich nicht an mich.
Она не вспоминает обо мне.
чаще перед обстоятельствами места с предлогами:
Er wohnt nicht in Spanien.
Он не живёт в Испании.
Sie arbeitet nicht in Dresden.
Она не работает в Дрездене.
Ich fahre nicht nach Berlin.
Я не поеду в Берлин.
Здесь может быть и частичное отрицание, что зависит он контекста или ударения:
Ich fahre nicht nach Berlin, sondern nach Paris.
Я поеду не в Берлин, а в Париж.
Ich fahre nicht nach Berlin. Ich bleibe lieber hier.
Я не поеду в Берлин. Я лучше останусь здесь.
В этом случае для показа однозначного частичного отрицания необходимо поставить предложную группу на первое место:
Nach Berlin fahre ich nicht.
В Берлин я не поеду.
чаще после обстоятельства времени с предлогом:
Ich schlief in der Nacht/die ganze Nacht/gestern nicht.
Я не спал ночью/всю ночь/вчера.
чаще после обстоятельства причины, цели, следствия, условия:
Er kommt wegen seiner Krankheit nicht.
Он не придёт из-за болезни.
всегда после наречий:
Das Spiel fand deswegen nicht statt.
Игра поэтому не состоялась.
чаще перед дополнением в Родительном падеже:
Die Besichtigung des Schlosses bedurfte nicht der Zustimmung des Besitzers.
Осмотр замка не требовал согласия его владельца.
перед именной частью сказуемого — перед существительным или прилагательным:
Er wird nicht Lehrer.
Он не станет учителем.
Sie wird nicht krank.
Она не заболеет.
Позиционно полное отрицание совпадает с частичным отрицанием.
Частичное отрицание здесь невозможно.
возможно перед дополнением, если оно распространённое – в целях лучшего понимания:
Er untersucht den Psychischen Zustand des Kranken nicht./Er untersucht nicht den Psychischen Zustand des Kranken.
Он не обследовал психическое состояние больного.
Место nicht в придаточном предложении:
Ich weiß, dass er nicht arbeitet.
Я знаю, что он не работает.
Ich weiß, dass er nicht Lehrer wird.
Я знаю, что не станет учителем.
Ich weiß, dass er den Freund nicht sieht.
Я знаю, что он не видит друга.
Ich weiß, dass er nicht an dich denkt.
Я знаю, что он не думает о тебе.
Ich weiß, dass sie das Buch nicht auf den Tisch legt.
Я знаю, что она не кладёт книгу на стол.
Особенность перевода конструкции nichtumhinkönnenzu+ инфинитив:
Ich kann nicht umhin, es zu tun.
Я не могу не делать этого.
Sie hat nicht umhingekonnt, das zu hören. – только в Перфекте!
Она не могла этого не слышать.
www.web-globus.de
Отрицание в немецком языке: nicht, kein, weder
Отрицание в немецком языке имеет одну особенность, делающую его принципиально отличным от русского языка. Отрицание может быть только одно единственное, двойное отрицание в этом языке не допускается.
Отрицание в немецком языке можно выразить с помощью отрицательных слов nicht, kein, weder … noch, nichts, niemand и так далее.
Пример:
Ist das dein Fahrrad? — Nein. Ist das dein Auto? — Ja. Ist das dein Fahrrad? — Nein, es ist nicht meins. Mein Fahrrad steht da drüben. Ist das dein Auto? — Ja, das ist mein Auto. Ist das nicht dein Fahrrad? — Nein. Ist das nicht dein Auto? — Doch. (Das ist mein Auto)
Вам следует в первую очередь запомнить разницу между словами nein и nicht:
nein – означает всегда «нет», а
nicht – «не».
При письме надо не забывать ставить запятую после слова „Nein“
Ist der Termin am Dienstag? Nein, der Termin ist erst am Donnerstag!
Отрицание с помощью nicht. Место nicht в предложении
Nicht может отрицать всё предложение, глагол или существительное с определенным артиклем.
Если в предложении один глагол, и мы его отрицаем, то nicht стоит в самом конце предложения перед точкой.
Arbeitest du? – Nein, ich arbeite nicht. Kochst du das Mittagessen? – Nein, ich koche das Mittagessen nicht. Kommst du mit uns ins Kino heute Abend? – Nein, ich komme mit euch in Kino heute Abend nicht.
Если в предложении 2 глагола (глаголы с отделяемыми приставками, предложения с модальными глаголами, инфинитив, прошедшее время), то nicht стоит на предпоследнем месте.
Macht sie die Tür zu? – Nein, sie macht die Tür nicht zu. Hast du heute die Zeitung gelesen? – Nein, die habe ich heute noch nicht gelesen. Muss ich alle Vokabeln lesen? – Nein, du musst alle Vokabeln nicht lesen, du musst sie lernen.
Если отрицаем предлог, то nicht стоит перед предлогом.
Fährst du mit dem Zug nach Lübeck? – Nein, ich fahre nicht mit dem Zug nach Lübeck, ich fahre mit dem Auto. Geht er morgens ins Schwimmbad? – Nein, er geht nicht ins Schwimmbad, er joggt im Park. Kommen Sie aus Frankreich? – Nein, ich komme nicht aus Frankreich.
Если предлог стоит на 1 месте, то nicht стоит в самом конце предложения.
Nicht не может стоять в начале предложения!
Fährst du mit diesem Zug nach Lübeck? – Nein, mit diesem fahre ich nicht. Geht er morgens ins Schwimmbad? – Nein, ins Schwimmbad geht er nicht. Kommen Sie aus Frankreich? – Nein, aus Frankreich komme ich nicht.
Nicht стоит перед отрицаемыми словами (сегодня, много, просто так, охотно и т.д.).
Liest du viel? – Nein, ich lese nicht viel. Trinkst du Mineralwasser? – Nein, ich trinke Mineralwasser nicht gern. Ich mache diese Aufgabe nicht heute.
Отрицание с помощью nicht
Часто нужно отрицать не все предложение, а только определенную часть или одно слово. В этом случае nicht будет стоять перед тем, что отрицаем. Интонацией сильно выделяем отрицание nicht и то, что отрицаем.
В некоторых случаях допустимо nicht в начале предложения. Если отрицаем какое-то слово или часть предложения, необходимо ввести альтернативу отрицанию (не сегодня, а завтра; не я, а он; не включить, а выключить и т.д.).
Для этого используется оборот nicht… , sondern.
Nicht Sonja hat das Glas gebrochen, sondern Christine. Du liest dieses Buch jetzt, nicht morgen. Nicht am Freitag, sondern am Samstag beginnt der Wettbewerb. Er konnte nicht ein Stück, sondern gleich eine ganze Torte essen. Wir gratulieren nicht nur dir, sondern deiner ganzen Familie. Bitte, schalte das Licht in dem Zimmer nicht aus, sondern ein.
Nicht может отрицать прилагательное, причастие или группу прилагательных. В этом случае nicht будет стоять перед прилагательным.
Mein Freund trägt oft dieses nicht gebügelte Hemd. Die nicht lange dauernde Vorlesung hat das Interesse der Studenten geweckt. Du hast mir ein noch nicht gelesenes Buch gegeben.
Отрицание с помощью kein
Существительное с определенным артиклем отрицается с помощью nicht.
Существительное с неопределенным артиклем отрицается с помощью kein-.
Существительное без артикля отрицается с помощью kein-.
Отрицательный артикль kein- склоняется точно так же, как и неопределенный артикль.
Во множественном числе нет неопределенного артикля, есть только отрицательный артикль keine.
Kasus
Maskulinum
Femininum
Neutrum
Plural
Nominativ
kein
keine
kein
keine
Akkusativ
keinen
keine
kein
keine
Dativ
keinem
keiner
keinem
keinen
Genitiv
keines
keiner
keines
keiner
Ist das ein Buch? – Nein, das ist kein Buch, sondern ein Heft. Ist das ein Radiergummi? – Nein, das ist kein Radiergummi, sondern ein Spitzer. Sind das _ Schüler? – Nein, das sind keine Schüler, sondern _ Studenten. (Plural !) Hat er eine Freundin? – Nein, er hat keine Freundin, er ist Single.
Если перед существительным стоит числительное eins, то оно склоняется как неопределенный артикль.
Числительное eins отрицается с помощью nicht.
Ich habe von meinen Eltern nicht ein Geschenk, sondern zwei. Helga hat Deutschland nicht einen Computer zu Hause, sondern drei. Meine Mutter hat nicht eine Bananentorte gebacken, sondern fünf.
Слово KEIN входит в число самых важных и часто употребляемых слов немецкого языка. Знакомимся с важными выражениями с этим словом.
20 важных выражений с KEIN
kein Wunder — неудивительно
auf keinen Fall — ни в коем случае
keine Zeit — нет времени
keine Ahnung haben — не иметь представления
keine Arbeit haben — быть безработным
keinen Sinn haben — не иметь смысла
keinen Anschluss bekommen — не дозвониться
auf keine Weise — никоим образом
auf ihn kein Verlass — на него нельзя положиться
du bist kein Kind mehr — ты уже не ребенок
ich habe kein Auge geschlossen — я не сомкнул глаз
ich habe kein Geld — у меня нет денег
er ist kein schlechter Mensch — он неплохой человек
keine Angst! — Не бойся!
keine Ursache — не за что
mach keine Witze! — не говори ерунды!
ohne Fleiß kein Preis — без труда не выловишь и рыбку из пруда
keinen Fitz wert sein — ничего не стоить
ich bekomme keine Luft — мне душно
machen Sie sich keine Mühe! — не беспокойтесь!
Отрицательные слова
положительно
отрицательно
Beispiele
Особа
jemand – кто-то
niemand – никто
Hast du da jemanden gesehen? — Nein, da habe ich niemanden gesehen.
Предмет
etwas, alles – что-то, все
nichts – ничего
Bestellst du etwas für sich? — Nein, ich bestelle nichts.
Время
jemals – когда-нибудь, oft – часто, immer – всегда, manchmal – иногда
nie, niemals – никогда
Wart ihr schon jemals in Österreich? — Nein, dort waren wir noch nie. In Österreich war ich niemals.
Место
irgendwo – где-то, überall – везде
nirgendwo, nirgends – нигде
Irgendwo in dem Flur liegt mein Regenschirm. Ich kann deine Brille nirgends finden.
Направление
irgendwohin – куда-то
nirgendwohin – никуда
Ich überlege mir, ob wir irgendwohin im Sommer in den Urlaub fahren. Mein Auto ist leider kaputt, ich kann jetzt nirgendwohin fahren.
Конструкции с отрицательным значением
«…ни …, ни … » («weder … noch»)
Tim kann nicht Deutsch sprechen. Er kann auch nicht Englisch sprechen. Tim kann weder Deutsch noch Englisch sprechen. – Тим не умеет говорить ни по-немецки, ни по-английски. Meine kleine Schwester kann noch nicht lesen. Sie kann auch nicht schreiben. Meine kleine Schwester kann weder lesen noch schreiben. – Моя маленькая сестра не может ни читать, ни писать.
не сделав что-то (ohne … zu)
Paul will reisen. Er will nicht viel Geld ausgeben.
Paul will reisen, ohne viel Geld auszugeben. – Пауль хочет путешествовать, не тратя много денег.
Sie geht weg. Sie verabschiedet sich nicht.
Sie geht weg, ohne sich zu verabschieden. – Она уходит, не прощаясь.
Предлоги с отрицательным значением
без + падеж Аккузатив (ohne + Akkusativ)
Wir beginnen die Feier. Wir warten auf dich nicht. Wir beginnen die Feier ohne dich. Der junge Mann fährt im Zug. Er hat keine Fahrkarte. Der junge Mann fährt im Zug ohne Fahrkarte.
кроме + падеж Датив (außer + Dativ)
Die ganze Touristengruppe ist pünktlich zum Bus gekommen, nur Herr Berger nicht. Die ganze Touristengruppe außer Herrn Berger ist pünktlich zum Bus gekommen. Meine Freunde haben schon alles in dieser Stadt gesehen, nur das Rathaus nicht. Meine Freunde haben alles in dieser Stadt außer dem Rathaus gesehen.
Правила русской орфографии и пунктуации онлайн. Полный академический справочник под ред. В.В. Лопатина.
На нашем сайте в электронном виде представлен справочник по орфографии русского языка. Справочником можно пользоваться онлайн, а можно бесплатно скачать на свой компьютер.
Надеемся, онлайн-справочник поможет вам изучить правописание и синтаксис русского языка!
РОССИЙСКАЯ АКАДЕМИЯ НАУК
Отделение историко-филологических наук Институт русского
языка им. В.В. Виноградова
ПРАВИЛА РУССКОЙ ОРФОГРАФИИ И ПУНКТУАЦИИ
ПОЛНЫЙ АКАДЕМИЧЕСКИЙ СПРАВОЧНИК
Авторы:
Н. С. Валгина, Н. А. Еськова, О. Е. Иванова, С. М. Кузьмина,
В. В. Лопатин, Л. К. Чельцова
Ответственный редактор В. В. Лопатин
Правила русской орфографии и пунктуации. Полный
академический справочник / Под ред. В.В. Лопатина. — М: АСТ, 2009. — 432 с.
ISBN 978-5-462-00930-3
Правила русской орфографии и пунктуации. Полный
академический справочник / Под ред. В.В. Лопатина. — М: Эксмо, 2009. — 480 с.
ISBN 978-5-699-18553-5
Справочник представляет собой новую редакцию
действующих «Правил русской орфографии и пунктуации», ориентирован на полноту
правил, современность языкового материала, учитывает существующую практику
письма.
Полный академический справочник предназначен
для самого широкого круга читателей.
Предлагаемый справочник подготовлен Институтом русского
языка им. В. В. Виноградова РАН и Орфографической комиссией при Отделении
историко-филологических наук Российской академии наук. Он является результатом
многолетней работы Орфографической комиссии, в состав которой входят лингвисты,
преподаватели вузов, методисты, учителя средней школы.
В работе комиссии, многократно обсуждавшей и одобрившей
текст справочника, приняли участие: канд. филол. наук Б. 3. Бук-чина, канд.
филол. наук, профессор Н. С. Валгина, учитель русского языка и литературы С. В.
Волков, доктор филол. наук, профессор В. П. Григорьев, доктор пед. наук,
профессор А. Д. Дейкина, канд. филол. наук, доцент Е. В. Джанджакова, канд.
филол. наук Н. А. Еськова, академик РАН А. А. Зализняк, канд. филол. наук О. Е.
Иванова, канд. филол. наук О. Е. Кармакова, доктор филол. наук, профессор Л. Л.
Касаткин, академик РАО В. Г. Костомаров, академик МАНПО и РАЕН О. А. Крылова,
доктор филол. наук, профессор Л. П. Крысин, доктор филол. наук С. М. Кузьмина,
доктор филол. наук, профессор О. В. Кукушкина, доктор филол. наук, профессор В.
В. Лопатин (председатель комиссии), учитель русского языка и литературы В. В.
Луховицкий, зав. лабораторией русского языка и литературы Московского института
повышения квалификации работников образования Н. А. Нефедова, канд. филол. наук
И. К. Сазонова, доктор филол. наук А. В. Суперанская, канд. филол. наук Л. К.
Чельцова, доктор филол. наук, профессор А. Д. Шмелев, доктор филол. наук,
профессор М. В. Шульга. Активное участие в обсуждении и редактировании текста
правил принимали недавно ушедшие из жизни члены комиссии: доктора филол. наук,
профессора В. Ф. Иванова, Б. С. Шварцкопф, Е. Н. Ширяев, кандидат филол. наук
Н. В. Соловьев.
Основной задачей этой работы была подготовка полного и
отвечающего современному состоянию русского языка текста правил русского
правописания. Действующие до сих пор «Правила русской орфографии и пунктуации»,
официально утвержденные в 1956 г., были первым общеобязательным сводом правил,
ликвидировавшим разнобой в правописании. Со времени их выхода прошло ровно
полвека, на их основе были созданы многочисленные пособия и методические
разработки. Естественно, что за это время в формулировках «Правил» обнаружился
ряд существенных пропусков и неточностей.
Неполнота «Правил» 1956 г. в большой степени объясняется изменениями, произошедшими в самом языке: появилось много новых слов и типов слов,
написание которых «Правилами» не регламентировано. Например, в современном
языке активизировались единицы, стоящие на грани между словом и частью слова;
среди них появились такие, как мини, макси, видео, аудио, медиа,
ретро и др. В «Правилах» 1956 г. нельзя найти ответ на вопрос, писать ли
такие единицы слитно со следующей частью слова или через дефис. Устарели многие
рекомендации по употреблению прописных букв. Нуждаются в уточнениях и
дополнениях правила пунктуации, отражающие стилистическое многообразие и
динамичность современной речи, особенно в массовой печати.
Таким образом, подготовленный текст правил русского
правописания не только отражает нормы, зафиксированные в «Правилах» 1956 г., но и во многих случаях дополняет и уточняет их с учетом современной практики письма.
Регламентируя правописание, данный справочник, естественно,
не может охватить и исчерпать все конкретные сложные случаи написания слов. В
этих случаях необходимо обращаться к орфографическим словарям. Наиболее полным
нормативным словарем является в настоящее время академический «Русский
орфографический словарь» (изд. 2-е, М., 2005), содержащий 180 тысяч слов.
Данный справочник по русскому правописанию предназначается
для преподавателей русского языка, редакционно-издательских работников, всех
пишущих по-русски.
Для облегчения пользования справочником текст правил
дополняется указателями слов и предметным указателем.
Составители приносят благодарность всем научным и
образовательным учреждениям, принявшим участие в обсуждении концепции и текста
правил русского правописания, составивших этот справочник.
Авторы
Ав. — Л.Авилова
Айт. — Ч. Айтматов
Акун. — Б. Акунин
Ам. — Н. Амосов
А. Меж. — А. Межиров
Ард. — В. Ардаматский
Ас. — Н. Асеев
Аст. — В. Астафьев
А. Т. — А. Н. Толстой
Ахм. — А. Ахматова
Ахмад. — Б. Ахмадулина
A. Цвет. — А. И. Цветаева
Багр. — Э. Багрицкий
Бар. — Е. А. Баратынский
Бек. — М. Бекетова
Бел. — В. Белов
Белин. — В. Г. Белинский
Бергг. — О. Берггольц
Бит. — А. Битов
Бл. — А. А. Блок
Бонд. — Ю. Бондарев
Б. П. — Б. Полевой
Б. Паст. — Б. Пастернак
Булг. — М. А. Булгаков
Бун. — И.А.Бунин
B. Бык. — В. Быков
Возн. — А. Вознесенский
Вороб. — К. Воробьев
Г. — Н. В. Гоголь
газ. — газета
Гарш. — В. М. Гаршин
Гейч. — С. Гейченко
Гил. — В. А. Гиляровский
Гонч. — И. А. Гончаров
Гр. — А. С. Грибоедов
Гран. — Д. Гранин
Грин — А. Грин
Дост. — Ф. М. Достоевский
Друн. — Ю. Друнина
Евт. — Е. Евтушенко
Е. П. — Е. Попов
Ес. — С. Есенин
журн. — журнал
Забол. — Н. Заболоцкий
Зал. — С. Залыгин
Зерн. — Р. Зернова
Зл. — С. Злобин
Инб. — В. Инбер
Ис — М. Исаковский
Кав. — В. Каверин
Каз. — Э. Казакевич
Кат. — В. Катаев
Кис. — Е. Киселева
Кор. — В. Г. Короленко
Крут. — С. Крутилин
Крыл. — И. А. Крылов
Купр. — А. И. Куприн
Л. — М. Ю. Лермонтов
Леон. — Л. Леонов
Лип. — В. Липатов
Лис. — К. Лисовский
Лих. — Д. С. Лихачев
Л. Кр. — Л. Крутикова
Л. Т. — Л. Н. Толстой
М. — В. Маяковский
Майк. — А. Майков
Мак. — В. Маканин
М. Г. — М. Горький
Мих. — С. Михалков
Наб. — В. В. Набоков
Нагиб. — Ю. Нагибин
Некр. — H.A. Некрасов
Н.Ил. — Н. Ильина
Н. Матв. — Н. Матвеева
Нов.-Пр. — А. Новиков-Прибой
Н. Остр. — H.A. Островский
Ок. — Б. Окуджава
Орл. — В. Орлов
П. — A.C. Пушкин
Пан. — В. Панова
Панф. — Ф. Панферов
Пауст. — К. Г. Паустовский
Пелев. — В. Пелевин
Пис. — А. Писемский
Плат. — А. П. Платонов
П. Нил. — П. Нилин
посл. — пословица
Пришв. — М. М. Пришвин
Расп. — В. Распутин
Рожд. — Р. Рождественский
Рыб. — А. Рыбаков
Сим. — К. Симонов
Сн. — И. Снегова
Сол. — В. Солоухин
Солж. — А. Солженицын
Ст. — К. Станюкович
Степ. — Т. Степанова
Сух. — В. Сухомлинский
Т. — И.С.Тургенев
Тв. — А. Твардовский
Тендр. — В. Тендряков
Ток. — В. Токарева
Триф. — Ю. Трифонов
Т. Толст. — Т. Толстая
Тын. — Ю. Н. Тынянов
Тютч. — Ф. И. Тютчев
Улиц. — Л. Улицкая
Уст. — Т. Устинова
Фад. — А. Фадеев
Фед. — К. Федин
Фурм. — Д. Фурманов
Цвет. — М. И. Цветаева
Ч.- А. П. Чехов
Чак. — А. Чаковский
Чив. — В. Чивилихин
Чуд. — М. Чудакова
Шол. — М. Шолохов
Шукш. — В. Шукшин
Щерб. — Г. Щербакова
Эр. — И.Эренбург
PAUTINI.RU — русскоязычные интернет сайты
Бесплатные онлайн игры по правописанию русского языка. Учителю на заметку — Дидактор
Встревоженность российского общества безграмотностью подрастающего поколения не могла не отразиться в интернет-пространстве. Здесь имеются не только ресурсы, банально подсказывающие лентяям, как правильно написать то или иное слово.
Предлагаю вашему вниманию несколько сервисов с онлайн тренажёрами, развивающими играми по русскому языку.
1. Проект «Уроки Русского языка», ставящий перед собой целью усиление внимания общественности к русскому языку, разместил онлайн тестер по следующим темам:
НЕ с разными частями речи
Правописание гласных в корне слова
Правописание суффиксов в глаголе и его формах
Правописание безударных личных окончаний глаголов
Пунктуация и орфография (4 теста)
Тестирование проводится незатейливо. Большинство из заданий представляют собой альтернативные тесты.
2. Создатели сайта для детей и взрослых «Лучшие игры» в разделе Головоломки предлагают свой тренажёр Правила правописания. Программа фиксирует ошибки, указывает количество правильных ответов, фиксирует время выполнения теста.
Тренажёр включает в себя подборку заданий на написание слов с суффиксами «ТСЯ» и «ТЬСЯ».
Правда, не мешало бы авторам тренажёра самим быть внимательнее в правописании:
3. Проект ИГРАЕМ САМИ среди большой коллекции развивающих игр по различным учебным предметам предлагает несколько онлайн игр и тренажёров по русскому языку по знанию пунктуации, букв русского алфавита, гласных и согласных букв и т.д.
В тестах используется технология перетаскивания объектов drag-and-drop, что придаёт большую динамику заданиям и предоставляет возможность использования их на интерактивной доске.
4. Сегодня в виртуальных магазинах для андроидов, наряду с приложениями развлекательного характера немало обучающих и развивающих игр, в том числе и по русскому языку. Известный разработчик приложений для мобильных устройств, компания Appscraft придумала игру «Орфография, игра-тест на знание русского языка».
Игру отличает хороший дизайн и большой спектр заданий.
Конечно, этим перечнем не исчерпываются подобные онлайн ресурсы. Однако общий их уровень подачи материала, универсальности большей частью пока не удовлетворяет запросам современного ученика.
Лучшие сайты для изучения русского языка онлайн
Неважно, сколько вам лет, но скорее всего, вам хочется говорить и писать грамотно. И несмотря на эпоху компьютеризации, которая принесла Т9 и другие системы автоматического исправления ошибок, настоящие знания не стали менее важными. Ведь, например, на государственных экзаменах автозамена текста уже не поможет.
Учиться сегодня можно без зубрёжки, а с интерактивом и лёгкостью. Существует множество сайтов для изучения русского языка, которые сделают процесс увлекательным.
Мы собрали 10 лучших сайтов по русскому языку для того, чтобы выучить теорию и попрактиковаться.
1. Грамота.ру
Пожалуй, самый популярный ресурс для тех, кто заинтересован в изучении русского языка. Чаще всего этот справочно-информационный портал используется для быстрой проверки правописания и проверки постановки ударения. Кроме того, на сайте можно изучать теорию — свод правил орфографии и пунктуации — и выполнять задания на усвоение материала.
2. Орфограммка
Сервис, позволяющий проверять целые тексты на соответствие нормам русского языка. Вы не только увидите конкретные ошибки (они будут выделены цветом), но и правила, связанные с каждой из них, и советы по улучшению текста.
3. Текстология
Сайт по русскому языку с полезной информацией не только для школьников, но и для филологов и лингвистов. Можно изучить все базовые языковые правила и найти много материалов как по русскому, так и по литературе.
4. Верные слова
Сайт для обучения русскому языку детей онлайн. Подойдёт для дошкольников и младшеклассников, которых мало привлекает бумажный учебник или скучные прописи. Красочный интерфейс и интерактивная платформа призваны заинтересовать ребёнка и расположить к изучению правописания. Подписка на сайт платная, есть свободный доступ к пробным урокам.
5. Интерактивный диктант
Классный ресурс для тех, кто хочет проверить свои силы и оценить уровень грамотности не выходя из дома. Некому начитывать текст? Не беда. Здесь нужно лишь вставлять буквы и знаки препинания в пропуски.
Каждый месяц к юбилею выдающегося русского писателя или поэта на сайте появляется новый диктант, основанный на произведениях юбиляра. Так, можно узнать, насколько вы грамотны, написав текст к 145-летнему юбилею Бунина или 125-летию со дня рождения Булгакова.
Сайт о русском языке создан при поддержке Городского методического центра Департамента образования Москвы.
6. Пишите живее!
Канцеляризмы — это штампы и обороты, характерные для официально-делового стиля. Часто они проникают в обычное неформальное общение, и речь становится нудной и неуклюжей.
Авторы сайта призывают избавляться от «мертвечины» (так канцеляриты называла известная переводчица Нора Галь) в письме и разговоре. Доступ бесплатный, а также есть возможность записаться на расширенную платную версию курса, где вы будете заниматься с преподавателем.
7. Веб-издание правил русского языка
Справочный сайт по изучению русского языка, созданный дизайнером и блогером Ильёй Бирманом, также известным своей типографской раскладкой. Главный плюс этого веб-издания — быстрый и удобный поиск, делающий обучение русскому языку онлайн более доступным. Все орфографические и пунктуационные правила хорошо структурированы, поэтому не составит труда найти нужную информацию.
8. Образование на русском
Несмотря на то что материал по большей части предназначается для иностранных граждан, изучающих русский язык, носители тоже найдут немало пользы. Можно углубить или освежить знания, а также попрактиковаться. Проект курируется Государственным институтом русского языка им. А. С. Пушкина.
9. Best-language
Ресурс, собравший все правила русского языка в одном месте. Фишкой сайта является краткость и лаконичность, которая позволяет запоминать большие объёмы информации и легко усваивать даже сложные правила. Хороший ресурс для общего повышения грамотности и подготовки к экзаменам.
10. Культура письменной речи
Тонны полезных материалов на сайте для всех, кто интересуется русским языком, — от обычных школьников до лингвистов и педагогов. Можно сфокусироваться на конкретных разделах — морфологии, стилистике, орфоэпии и так далее, изучать примеры сочинений и изложений, и даже участвовать в конкурсах и проходить интересные тесты на эрудицию. Есть раздел с типичными ошибками, с которым рекомендуется ознакомиться каждому, кто хочет писать грамотно.
Курсы экстерната и домашней школы «Фоксфорда»
Но что, если необходима комплексная подготовка? Иногда разрозненных образовательных сайтов по русскому языку недостаточно. Если вы чувствуете потребность в тщательной проработке пробелов в знаниях, приходите на базовые курсы русского языка от домашней онлайн-школы «Фоксфорда».
Учёба проходит удалённо, не нужно тратить время на дорогу или самостоятельно собирать информацию по крупицам с разных сайтов про русский язык для школьников. Весь материал тщательно структурирован и разложен по полочкам.
Здесь преподают педагоги из ведущих вузов России — кандидаты филологических наук, авторы учебно-методических пособий и составители задач таких олимпиад, как, например, «Русский медвежонок».
Видеоуроки можно смотреть с любого устройства, включая планшет или телефон. Каждый урок сопровождают интерактивные задания.
Есть крутая функция абонементов. Абонемент включает в себя базовые предметы школьной программы по классам — курсы за целый год обучения.
Можно заниматься в своём ритме: просто смотрите курсы в записи, когда захочется. Замедляйте и ускоряйте воспроизведение, ставьте на паузу и просматривайте видео сколько угодно раз — полная свобода!
Пишите грамотно!
Проверка орфографии и грамматики в Office
Все приложения Microsoft Office поддерживают проверку орфографии, и большинство из них поддерживает проверку грамматики.
Если проверка правописания работает не так, как вы ожидали, см. статью Средство проверки орфографии и грамматики помечает текст, который не должен быть помечен. Если вам нужно выполнить проверку правописания на языке, отличном от языка по умолчанию, см. статью Проверка орфографии и грамматики на другом языке.
Используете Office 365? Возможно, вас заинтересует новая функция Word — «Корректор»! Подробные сведения см. в статье Корректор.
Запуск средства проверки орфографии и грамматики вручную
Чтобы запустить проверку правописания в файле, нажмите клавишу F7 или выполните следующие действия:
Откройте приложение Office и перейдите на вкладку Рецензирование. В Access и InfoPath можно пропустить это действие. В Project перейдите на вкладку Проект.
Нажмите кнопку Орфография или Правописание.
Если программа обнаружит орфографические ошибки, появится диалоговое окно с первым из слов с ошибками, найденных средством проверки орфографии.
После того как вы примете решение по ошибке (пропустить ее, добавить слово в словарь или изменить его), приложение перейдет к следующему неправильно написанному слову.
Щелкните заголовок ниже, чтобы получить дополнительные сведения.
В большинстве приложений Office по мере ввода текста выполняется автоматическая проверка правописания, поэтому вы сразу можете увидеть ошибки во время работы.
Примечания:
Автоматическая проверка орфографии и грамматики отсутствует в Access, Excel и Project. Можно вручную запустить проверку орфографии, нажав клавишу F7.
Автоматическая проверка грамматики доступна только в Outlook, Word и PowerPoint 2013 (или более новой версии).
Офис отмечает потенциальные орфографические ошибки красной волнистой линией, а потенциальные грамматические ошибки отмечены синей волнистой линией.
Если орфографические или грамматические ошибки не помечаются, автоматическая проверка может быть отключена. Можно включить автоматическую проверку правописания.
Если вы видите орфографическую или грамматическую ошибку и вам нужна помощь, чтобы ее исправить, щелкните подчеркнутое слово или фразу правой кнопкой мыши и выберите один из предложенных вариантов.
Если в приложении Office слово отмечено, как содержащее ошибку, но вы написали его правильно, выберите пункт Добавить в словарь, чтобы в будущем это слово не отмечалось как неправильное. Дополнительные сведения см. в статье Добавление слов в словарь проверки орфографии и их изменение.
Если вы не хотите, чтобы в процессе работы приложение Office помечало возможные ошибки волнистыми линиями, вы можете отключить автоматическую проверку правописания.
Откройте параметры проверки правописания:
В OneNote, PowerPoint, Publisher, Visio и Word: в меню Файл выберите пункт Параметры и щелкните Правописание.
В InfoPath: на вкладке Главная щелкните стрелку рядом с надписью Орфография и выберите команду Параметры проверки орфографии.
В Outlook: в меню Файл щелкните Параметры, выберите Почта и нажмите кнопку Орфография и автозамена.
Установите или снимите флажок Проверять орфографию в процессе набора текста. Кроме того, в приложениях с автоматической проверкой грамматики можно установить или снять флажок Отмечать грамматические ошибки в процессе набора текста.
Примечание: В Word можно включать и отключать средство проверки орфографии для документа, с которым вы работаете, или для всех новых документов. Выберите значение в списке Исключения, а затем установите или снимите флажки Скрыть орфографические ошибки только в этом документе и Скрыть грамматические ошибки только в этом документе.
Если вы не хотите, чтобы в приложении Office проверялась грамматика (ни при запуске проверки правописания, ни автоматически по мере ввода), эту функцию можно отключить.
Откройте параметры проверки правописания:
В OneNote, PowerPoint, Publisher, Visio и Word: в меню Файл выберите пункт Параметры и щелкните Правописание.
В InfoPath: на вкладке Главная щелкните стрелку рядом с надписью Орфография и выберите команду Параметры проверки орфографии.
В Outlook: в меню Файл щелкните Параметры, выберите Почта и нажмите кнопку Орфография и автозамена.
Снимите флажки Автоматически проверять грамматику и Также проверять грамматику.
Примечание: не во всех приложениях Office присутствуют оба эти параметра.
В Word, Outlook, PowerPoint 2013 (или более новой версии) можно принудительно выполнить повторную проверку ранее пропущенных слов и выражений.
Откройте документ или элемент, который вы хотите проверить.
Откройте вкладку Файл и выберите команды Параметры > Правописание > Повторная проверка. В Outlook нужно выбрать пункты Файл > Параметры > Почта и нажать кнопку Орфография и автозамена.
Нажмите кнопку Да, когда появится предупреждение о сбросе параметров проверки орфографии и грамматики.
Нажмите кнопку ОК в диалоговом окне, чтобы вернуться к документу, а затем еще раз запустите проверку орфографии и грамматики.
Нам важно ваше мнение
Эта статья последний раз была обновлена Беном28 октября 2019 г. Если она оказалась полезной для вас (и тем более, если нет), нажмите соответствующую кнопку ниже и поделитесь своими идеями о том, как ее улучшить.
См. также
Проверка удобочитаемости документа
Средство проверки орфографии и грамматики помечает текст, который не должен быть помечен
Выбор параметров грамматики и стиля письма в Office 2013 и более ранних версий
Добавление слов в словарь проверки орфографии
Средство проверки правописания неправильно работает с другим языком
Настройка автозамены: написание прописными буквами, правописание и символы
Проверка орфографии перед отправкой сообщения в Outlook
Приложения Office для Mac автоматически проверяют наличие возможных орфографических и грамматических ошибок при вводе текста. Если вы предпочитаете проверять орфографию и грамматику уже в готовом документе, отключите автоматическую проверку или можно одновременно выполнить проверку орфографии и грамматики.
Word
Автоматическая проверка орфографии и грамматики при вводе
Слово помечает потенциальные орфографические ошибки красной волнистой линией, а потенциальные грамматические ошибки отмечены зеленой волнистой линией.
Совет: Если орфографические и грамматические ошибки не помечаются, вероятно, вам нужно включить автоматическую проверку правописания, о которой пойдет речь в следующей процедуре.
Если вы видите орфографическую и грамматическую ошибку, удерживая нажатой клавишу CONTROL, щелкните слово или фразу и выберите один из вариантов.
Если приложение Word неправильно пометило слово как опечатку и вы хотите добавить это слово в словарь, чтобы приложение Word правильно распознавало его в дальнейшем, см. раздел Добавление слов в словарь проверки орфографии и их изменение
В меню Word выберите Параметры > Правописание.
В диалоговом окне Правописание в разделе Орфография установите или снимите флажок Автоматически проверять орфографию.
В разделе Грамматика установите или снимите флажок Автоматически проверять грамматику.
Закройте диалоговое окно, чтобы сохранить изменения.
На вкладке Рецензирование нажмите кнопку Правописание.
Если Word находит возможную ошибку, открывается диалоговое окно Правописание, в котором орфографические ошибки выделяются красным цветом, а грамматические — зеленым.
Чтобы устранить ошибку, выполните одно из указанных ниже действий.
Введите исправление в соответствующем поле и нажмите кнопку Изменить.
В разделе Варианты выберите нужное слово и нажмите кнопку Изменить.
Чтобы пропустить ошибку, выполните одно из указанных ниже действий.
Чтобы пропустить только этот экземпляр ошибки, нажмите кнопку Пропустить.
Чтобы пропустить все экземпляры ошибки, нажмите кнопку Пропустить все.
Для грамматической ошибки щелкните Следующее предложение, чтобы пропустить экземпляр этой ошибки и перейти к следующей.
Если нужно пропускать слово с ошибкой во всех документах, нажмите кнопку Добавить, чтобы добавить слово в словарь. Это применимо только для слов с орфографическими ошибками. Свою грамматику вы не можете добавить в словарь.
После исправления или пропуска ошибки Word переходит к следующей. По окончании проверки документа в Word появляется сообщение о том, что проверка правописания завершена.
Нажмите кнопку ОК, чтобы вернуться к документу.
Список пропускаемых слов и грамматических ошибок можно очистить, после чего приложение Word снова проверит ошибки в орфографии и грамматике, которые вы до этого решили пропустить.
Примечание: Список пропускаемых слов и грамматики сбрасывается только для документа, который открыт в данный момент. Это действие на затрагивает орфографические и грамматические ошибки, которые вы решили пропустить в других документах Word.
Откройте документ, который необходимо проверить.
В меню Сервис наведите указатель на пункт Правописание и выберите Сбросить пропускаемые слова и грамматику.
Word предупредит вас о том, что эта операция приведет к сбросу средств проверки орфографии и грамматики.
Нажмите кнопку Да, чтобы продолжить.
Откройте вкладку Рецензирование и щелкните Правописание, чтобы проверить орфографию и грамматику.
Outlook
Автоматическая проверка орфографии и грамматики при вводе
По умолчанию приложение Outlook проверяет орфографию в процессе ввода текста. В Outlook используется красная пунктирная линия подчеркивания для обозначения возможных орфографических ошибок, а также зеленая пунктирная линия для обозначения возможных грамматических ошибок.
Если вы видите слово с пунктирной линией подчеркивания, то удерживая нажатой клавишу Control, щелкните слово или фразу и выберите один из вариантов.
В контекстном меню выполните одно из указанных ниже действий.
Выберите один из предлагаемых вариантов в верхней части контекстного меню.
Нажмите кнопку Пропустить правописание, чтобы пропустить одно вхождение слова.
Чтобы добавить слово в орфографический словарь, щелкните Добавить в словарь.
Открыв сообщение электронной почты, выполните указанные ниже действия.
Чтобы Outlook автоматически выполнял исправление орфографических ошибок, в меню Outlook выберите пункт Параметры. В разделе Личные параметры щелкните Правописание. Щелкните в поле рядом сПроверкаорфографии при вводе текста.
Чтобы включить или отключить автоматическую проверку грамматики, в меню Outlook выберите пункт Параметры. В разделе Личные параметры щелкните Правописание. Щелкните в поле рядом с пунктом Проверка грамматики при вводе.
Вы можете исправить орфографические и грамматические ошибки во всем тексте после создания сообщения или другого элементы.
В меню Правка наведите указатель на пункт Правописание и выберите Правописание.
Выполните любое из описанных ниже действий.
В списке вариантов выберите нужное слово или введите новый вариант правописания в поле в верхней части, и нажмите Изменить.
Нажмите Пропустить, чтобы пропустить это слово и перейти к следующему слову с ошибкой.
Чтобы добавить слово в орфографический словарь, щелкните Добавить.
Совет: Для пропуска слова и перехода к следующей ошибке правописания используется сочетание клавиш +;.
PowerPoint
В PowerPoint можно проверять орфографию, но не грамматику.
Автоматическая проверка орфографии при вводе
PowerPoint автоматически проверяет и отмечает потенциальные орфографические ошибки с волнистым красным подчеркиванием.
Совет: Если орфографические ошибки не помечаются, вероятно, вам нужно включить автоматическую проверку орфографии, о которой пойдет речь в следующей процедуре.
Если вы увидите орфографическую ошибку, щелкните слово или фразу правой кнопкой мыши (или левой, удерживая клавишу CTRL) и выберите один из предложенных вариантов.
В меню PowerPoint выберите разделы Параметры > Проверка орфографии.
В диалоговом окне Проверка орфографии установите или снимите флажок Автоматически проверять орфографию.
На вкладке Рецензирование нажмите кнопку Проверка орфографии.
При обнаружении ошибки откроется область Проверка орфографии с возможными вариантами исправления.
Чтобы устранить ошибку, выполните одно из указанных ниже действий.
Исправьте ошибку на слайде.
Выберите одно из слов, предложенных в области Проверка орфографии, а затем нажмите кнопку Изменить.
Чтобы пропустить ошибку, выполните одно из указанных ниже действий.
Чтобы пропустить только этот экземпляр ошибки, нажмите кнопку Пропустить.
Чтобы пропустить все экземпляры ошибки, нажмите кнопку Пропустить все.
Чтобы пропускать ошибку во всех документах и добавить слово в словарь, нажмите кнопку Добавить.
После исправления, игнорирования или пропуска PowerPoint перейдет к следующей ошибке. После завершения проверки презентации в PowerPoint появится сообщение о том, что проверка орфографии закончена.
Нажмите кнопку ОК, чтобы вернуться к презентации.
Excel
В Excel можно проверять орфографию, но не грамматику.
Проверка орфографии во всей презентации
На вкладке Рецензирование нажмите кнопку Проверка орфографии.
Примечание: Диалоговое окно Орфография не откроется, если ошибки правописания не обнаружены или вы пытаетесь добавить слово, которое уже есть в словаре.
Выполните любое из описанных ниже действий.
Задача
Необходимые действия
Замена слова
В разделе Варианты выберите нужное слово и нажмите кнопку Изменить.
Изменение каждого экземпляра этого слова в данном документе
В разделе Варианты выберите нужное слово и нажмите кнопку Изменить все.
Пропуск слова и переход к следующему слову с ошибкой
Нажмите кнопку Пропустить.
Пропуск каждого экземпляра этого слова в данном документе и переход к следующему слову с ошибкой
Нажмите Пропустить все.
Дополнительные материалы
Проверка орфографии и грамматики на другом языке
Способы проверки орфографии онлайн | Юзабилити
Русский язык знают все русские люди, но некоторые даже не умеют правильно говорить на нем, а уж тем более – писать. Именно поэтому всем необходимо знать, как можно быстро и качественно проверить орфографию в написанном тексте.
Многие студенты выполняют свои работы, печатая. Также в наши дни немало копирайтеров трудятся в интернете, набирая статьи и отправляя их заказчикам. Этим людям знать, как возможна проверка орфографии онлайн, просто необходимо.
Грамота.ру – очень популярный и многим известный сайт, с помощью которого можно изучать русский язык, постоянно совершенствовать навыки письма, а также выполнять упражнения, чтобы заучить недопонятые правила.
На этом сервисе присутствует возможность проверки орфографии онлайн по ссылке http://gramota.ru/spravka/orfo. Также на портале можно задать вопрос справочному бюро про какое-нибудь правило русского языка или, например, на тему пунктуации. Сделать это можно на главной странице, как и проверить написание какого-либо слова.
Выполняет проверку орфографии онлайн и Яндекс. Он позволяет проверить орфографию текста на своем или чужом сайте. Такой способ очень удобен для тех, кому нужно провести проверку статей в своем блоге, например.
Для этого на страничке Яндекс.Вебмастера необходимо выбрать «Проверить орфографию» и ввести в появившейся строке адрес проверяемой страницы сайта. Через несколько мгновений Яндекс выдаст результат, показав страницу сайта с текстом, ошибки в котором будут выделяться желтым фоном. Конечно, сервис этот очень мощный, но и он, как многие другие, некоторые слова может просто-напросто не знать.
Также по ссылке http://api.yandex.ru/speller/ можно найти сервис, позволяющий произвести проверку правописания текста онлайн, созданный для вебмастеров и разработчиков. Любой желающий может подключить такую проверку к своему сайту по размещенной на странице инструкции. Работу проверки возможно тут же проверить, введя текст на русском, английском или украинском языках.
Сервис Адвего позволяет провести мультиязычную проверку орфографии онлайн. Русский язык стоит по умолчанию, но если нужно, можно установить другой, один из множества предлагаемых сайтом. Также пользователи сервиса могут применить семантический анализ текста и проверить его уникальность.
Для многих Адвего – лучший ресурс, так как на нем еще можно и заработать на написании статей, и заказать статьи у копирайтеров, и много чего другого. Производить проверку орфографии онлайн на сайте очень удобно, а результат радует своим качеством.
Текст необходимо вставить в окошко и нажать на кнопку «Проверить». Если язык не русский, следует поменять его в поле над окном для текста.
Затем на экране появятся результаты проверки, в них можно посмотреть возможные ошибки в тексте, количество символов и слов, а также другие детали анализа, которые будут интересны человеку, пишущему тексты на заказ.
Возможна проверка правописания текста онлайн еще на одном сайте — http://orthography.morphology.ru/. Ничего лишнего на нем вы не найдете, понадобится лишь вставить нужный текст в поле под словами «Проверка орфографии» и нажать на кнопку Check.
Через секунду вы увидите результат, в котором ошибочные слова будут подчеркнуты красной волнистой линией. Также на этом сайте присутствует полезный морфологический словарь, он может пригодиться тем, кто плохо разбирается в правописании.
К сожалению, в интернете нет такого ресурса, которым выполняется проверка пунктуации онлайн. Возможно, кто-то и старался создать его, но это невозможно. Дело в том, что расстановка знаков препинания в русском языке может быть довольно сложной и не всегда понятной. Согласитесь, часто необходимо знать смысл предложения, чтобы решить, где должна стоять запятая.
Вспомним известную фразу «Казнить нельзя помиловать», в которой от знака препинания зависит не просто смысл а предложения, а еще и сохранность жизни человека. Этот пример говорит о том, что расстановка запятых очень важна, а компьютер не в состоянии самостоятельно распознать смысл текста.
Когда пишешь электронные письма, очень удобно знать, где совершил ошибку сразу, чтобы не копировать текст и не переносить его в сервис, который проверяет правописание онлайн. В этом случае очень пригодятся встроенные в браузер модули и плагины. В Opera, Google Chrome и Mozilla Firefox проверка встроена прямо в браузер, а в Internet Explorer необходимо установить плагин IE7pro.
Проверка орфографии в браузере Mozilla Firefox
В браузере уже есть модуль проверки, поэтому нужно установить только словарь необходимого языка. По умолчанию установлен английский, а чтобы установить другой, надо открыть контекстное меню браузера правым щелчком мыши и выбрать в нем пункт «Проверка правописания«, затем активировать его, снова открыть меню и выбрать в пункте «Язык» подпункт под названием «Добавить словарь«.
Далее потребуется лишь выбрать нужный язык из списка и установить его. Для этого нужно пройти по предложенной ссылке, а там уже будет кнопка «Добавить в Firefox«. В дальнейшем последует обычная процедура, как при установке любого дополнения для этого браузера.
Проверка орфографии в браузере Opera
В браузере Opera 12 онлайн проверка правописания производится автоматически, если же этого не происходит, необходимо разобраться, в чем проблема. Первым делом убедитесь, что дело не в настройках сайта. Если сайт не при чем, проверьте, загружен ли словарь орфографии русского языка в браузер. Для этого нужно открыть контекстное меню и выбрать пункт «Словари«, затем подпункт «Добавить/Удалить словарь» и выбрать в списке нужный язык.
Проверка орфографии в браузере Google Chrome
В Google Chrome имеется встроенный инструмент проверки правописания, автоматически проверяющий все текстовые поля и веб-формы. В настройках вы можете выбрать необходимый язык и, если необходимо, отключить автоматическую проверку орфографии.
Проверка орфографии в браузере Internet Explorer
Вам следует установить плагин IE7pro, так как в нем есть модуль проверки Spell Check. После того как вы загрузите его на компьютер и установите, настройте модуль проверки орфографии. Чтобы активировать модуль, зайдите в настройки плагина и отметьте его галочкой в пункте «Modules«. Настройки сохранятся, и впредь проверка орфографии будет автоматической, как и в других браузерах.
Как видите, если вас интересует проверка орфографии и пунктуации онлайн, в вашем распоряжении немалое количество способов, к которым можно прибегнуть. Вам остается лишь выбрать наиболее подходящий из них. Если вас не устраивает проверка браузера или вы хотите удостовериться в ее точности, вы всегда можете воспользоваться одним из приведенных выше сервисов.
ТОП-6 сервисов и приложений для изучения орфографии и пунктуации русского языка: marjulia — LiveJournal
Использованы материалы сайтов Лайфхакер и Theory&Practice
1. Грамота.ру
На сайте Грамота.ру, помимо словарей и справочного бюро, в разделе «Класс» вы найдёте пособия по русскому языку. Там же обратите внимание на раздел «Репетитор онлайн», состоящий из двух частей «Интерактивные диктанты» и «Учебник ГРАМОТЫ».
https://lifehacker.ru/izuchenie-russkogo-yazyka/
Мобильное приложение Грамота.ру для Android. Проходите мини-диктанты, изучайте правила пунктуации и орфографии! Список диктантов постоянно пополняется диктантами по произведениям классиков отечественной и мировой литературы.
2. Тотальный диктант
На сайте «Тотального диктанта» собраны все основные правила и разобраны трудности русского языка. Там вы найдёте онлайн-курс Тотального диктанта, курс по истории русской орфографии и коллекцию диктантов. В любое время вы можете проверить себя и написать диктант онлайн. К каждому диктанту есть аудио, а результат проверяется автоматически.
3. Интерактивный диктант
На этом ресурсе Городского методического центра Департамента образования Москвы появляются диктанты, основанные на произведениях выдающихся русских писателей. В каждом тексте есть пропуски — вам нужно вставлять недостающие символы, выбирая их из предложенных вариантов. По завершении система подсчитывает количество допущенных ошибок и выдаёт результат.
https://lifehacker.ru/izuchenie-russkogo-yazyka/
4. Русский язык — грамотей
Внутри этой программы более 16 000 тестовых заданий по орфографии, пунктуации и другим разделам. Как утверждает разработчик, в составлении вопросов принимали участие специалисты по русскому языку. Столкнувшись с трудным тестом, вы можете запросить помощь — программа отобразит связанный с вопросом теоретический раздел.
Приложение включает тестовые задания по русскому языку в рамках школьной программы. Режим Тренировка позволяет выполнять задания по вариантам, по темам и по случайным вопросам, а также изучить теоретический материал по каждой теме.
5. «Орфография»
iOS,Google play
«Орфография» представляет собой короткие карточки с текстами из литературных произведений, куда нужно вставить пропущенные буквы, — точно как в школьном учебнике русского языка. После выполнения можно кликнуть на любое из заданий и прочитать теорию. Упражнения простые, на уровне пятого-шестого классов, поэтому приложение в первую очередь подойдет школьникам. Бесплатно доступны только 25 карточек.
С помощью этой программы вы можете проверить и, если необходимо, подтянуть знания пунктуации. Работает она следующим образом: вам показывают предложения из известных книг, а вы расставляете знаки препинания. Система оценивает результат и комментирует ошибки. Бесплатно можно обработать лишь 20 предложений, ещё 200 доступны за символическую цену.
https://lifehacker.ru/izuchenie-russkogo-yazyka/
Ещё о книгах по русской грамматике, а также орфографии и пунктуации смотрите здесь: https://marjulia.livejournal.com/298784.html
Онлайн проверка орфографии. Где можно проверить текст на ошибки?
Многим блоггерам близка проблема с орфографией. Это не мудрено, многие из нас проводили большую часть свободного времени за компьютерными играми, на форумах и порталах на подобии Упячки. Вследствие чего выработалась своеобразная форма общения, стиль общения, которые очень далеки от норм русского языка. И теперь у тех, чья работа связанна с написанием текстов, возникают определенные трудности с этим делом. Появляется необходимость в проверке орфографии и исправлении текста.
Да и в принципе, любой человек, работающий с большими объемами материала, не застрахован от ошибок и опечаток. И время от времени будет совершать грамматические и пунктуационные ошибки.
А ошибки в нашем деле непозволительны, так как их наличие может пагубно повлиять на формирование постоянной аудитории читателей. Для большинства посетителей это очень весомый фактор, способный их отпугнуть.
Поэтому рассмотрим несколько инструментов и онлайн сервисов для проверки написания слов.
Онлайн проверка орфографии
Яндекс Спеллер
Яндекс Спеллер позволяет проверить правописание на 3-х языках: русском, английском и украинском. Работа Спеллера основывается на использовании орфографического словаря. Более того, этот словарь включает в себя множество современных слов и постоянно пополняется.
Но, кроме стандартных возможностей, у Спеллера есть 2 особые:
Подключить проверку орфографии на свой сайт, настроить стили оформления. Разве плохо иметь свою собственную онлайн проверку правописания? Это задержит посетителей на сайте, что не может не радовать. И тем более, это может сформировать постоянную аудиторию, которая будет пользоваться онлайн проверкой орфографии на Вашем сайте. Работу можно посмотреть ниже.
Проверка правописания онлайн
Проверить
Параметры… Инструкция по подключению Яндекс Спеллера на своем сайте.
Очень часто, при написании статьи, бывают затруднения с написание какого–нибудь слова. Поэтому, изначально я писал в Word, а уже потом добавлял на сайт. Но у этого метода было 2 очень неудобных для меня момента: Яндекс спеллер имеет куда больший словарь, с кучей современных слов, в отличие от Ворда. И в цепи Ворд – редактор – сайт присутствовало лишнее звено, которое можно было достаточно легко отцепить с помощью подключения api Yandex Спеллера непосредственно к редактору WordPress.
Подключить можно с помощью плагинов. Устанавливать любой на выбор:
Q2W3 Yandex Speller
Yandex Speller Application
Установить их можно и из панели администратора WordPress.
Но MS Word не стоит откидывать, так как это одна из лучших программ проверки пунктуации, сайты онлайн проверки пунктуации определенно ей уступают.
Проверка орфографии на сайте от Яндекса
Проверка орфографии сайта. Если текст уже опубликован на сайте, то правописание текста можно проверить с помощью онлайн сервиса проверки правописания от Яндекса:
http://webmaster.yandex.ua/spellcheck.xml.
Достаточно просто ввести Url и все слова с ошибками будут выделены желтым цветом.
Как и Спеллер, сервис проверки орфографии основывается на работе орфографического словаря с большим количеством современных слов.
Сервис Адвего — онлайн проверка орфографии
Одна из лучших бирж контента, являющаяся посредником между теми, кто продает статьи и теми, кто их покупает, делает все, для того, чтобы работающие с ней чувствовали себя комфортно. В помощь, биржа предоставляет возможность проверять текст на уникальность, делать СЕО-анализ текста, а также проверять на ошибки статьи.
Ссылка на сервис проверки от Advego.
Как видно из скрина, словарь у биржи контента такой себе. Даже немножко удивляет, когда биржа копирайтинга не внесла слово «копирайтинг» в свой словарь.
Но, для поверхностной проверки вполне подойдет. Кроме того, есть ряд полезных инструментов, позволяющих сделать анализ текста.
тестов и викторин по русскому языку
Испытания России Сборник развлекательных и познавательных тестов о России, русском языке, людях, истории и культуре.
Тест по русскому языку онлайн Ответьте на 50 вопросов разной сложности с несколькими вариантами ответов для оценки
ваш уровень русского. Этот тест предоставляется по языковой ссылке
школа.
Русский
Грамматические упражнения Проверьте свои знания русского языка с помощью этих упражнений на регистр, алфавит, правила орфографии и т. Д.
транслитерация и спряжение глаголов.
Англо-русский словарный запас Эти тесты помогут вам выучить и пересмотреть словарный запас. Они
охватывают такие темы, как числа, мебель, время, тело, работа и
занятия, еда и красители.
Онлайн-тест по русскому языку Хороший ресурс для изучения базового словарного запаса русского языка в викторине.
формат. Включает тесты по таким предметам, как числа, мебель, время,
тело, работа и занятия, еда и красители.
Упражнения Голоса Этот сайт может похвастаться множеством дополнительных упражнений к книге Голоса Георгия Митревского. Хотя владение книгой поможет
ваша грамматика, вам не нужно ее иметь, чтобы сдавать тесты и учить
новая русская лексика и грамматика.
Языковой центр Liden & Denz Испытайте себя с 65 грамматическими вопросами от начального уровня (A1) до более низкого продвинутого (B2 +). Поставляется языковыми центрами Liden & Denz в Санкт-Петербурге.
Взаимодействие с другими людьми
Авторские права 2001-2021 MasterRussian.com | Конфиденциальность
Политика | Свяжитесь с нами
26 лучших (и худших) онлайн-курсов русского языка в 2021 году
Найти лучших онлайн-курсов русского языка может быть сложно для большинства учащихся.
Параметры контента только немного улучшились с тех пор, как я выучил русский несколько лет назад, так что еще есть возможности для гораздо большего. 🙂
Русский, безусловно, один из наименее популярных основных иностранных языков в мире, но из всех языков, которые я выучил, он был, безусловно, одним из самых полезных.
Если вы только начинаете, позвольте мне заверить вас, что вы приняли отличное решение.
Есть много веских причин изучать русский язык — поездка в Россию того стоит, но она также открывает множество возможностей для бизнеса и карьерного роста во многих сферах.
Для тех, кто живет в США, в некоторых частях страны есть огромные русские анклавы, поэтому знание русского языка позволяет вам общаться со своими соседями в этих местах.
Кроме того, это действительно интересный язык с богатой историей. 🙂
Прочтите мои предыдущие сообщения о моем опыте погружения в Россию.
Итак, сегодня я поделюсь с вами лучшими (и худшими) из всех популярных онлайн-курсов русского языка (у меня была возможность использовать большинство из этих курсов).
Я выскажу свое обоснованное мнение как свободно говорящего / изучающего русский язык.
Ниже вы найдете плюсы и минусы каждого курса, цены и краткое описание. Если на этом сайте есть отзыв о продукте, я сделаю ссылку на него.
Некоторые из них связаны партнерской программой, а большинство — нет.
ПРИМЕЧАНИЕ. Некоторые из перечисленных ниже предметов, вероятно, не обязательно являются «курсами» для русского языка как такового. Я включил их потому, что они достаточно популярные российские инструменты, чтобы их можно было включить.
ОТКАЗ ОТ ОТВЕТСТВЕННОСТИ: Приведенные ниже комментарии являются личным мнением.
Лучшие курсы русского языка онлайн (самые популярные русские ресурсы)
1. Rocket Russian
Стоимость: От 99,95 долларов США (автоматически применяется скидка)
Резюме: Я всегда был ярым поклонником серии Rocket Languages, и русская версия, безусловно, моя главная рекомендация. Из всех вариантов курса русского, перечисленных ниже, Rocket Russian больше всего подходит ученикам структурированного курса , поскольку он разработан для линейной прогрессии.
Хорошие новости для тех, кто хочет идти своим путем: вы не обязаны идти по их пути и можете пропускать уроки, если хотите.
Аудио-уроки в основном представляют собой формат в стиле подкастов, за которым очень легко следовать, без дополнительных материалов и аудио-диалогов, которые дают вам только необходимые русские диалоги и ничего более (что мне нравится). Курс Rocket Russian очень хорошо охватывает все языковые навыки, а встроенное распознавание голоса очень точное, если это то, что вы сочтете полезным (в нем используется технология Google Web Speech).
Для структурированных учащихся Rocket — отличный курс русского языка.
Вы также можете ознакомиться с моим обзором Rocket Russian.
Что мне в нем нравится:
Для учащихся, желающих структурировать свой курс обучения (хотя и не ограниченных этим), он имеет очень четкую линейную прогрессию.
Он равномерно охватывает все 4 основных навыка (аудирование, говорение, чтение, письмо).
Должен предлагать уровни 2 и 3 для учащихся более высокого уровня.
УНИКАЛЬНОЕ ПРЕДЛОЖЕНИЕ:
Присоединяйтесь к Гильдии, нажав здесь, выберите русский язык, и я пришлю вам уникальное предложение для Rocket Russian, эксклюзивное только для моих читателей.
2. Русский Под101
Стоимость: Начинается от 4 долларов в месяц.
Резюме: RussianPod101 в последние годы добился больших успехов. Раньше это была беспорядочная мешанина аудио и видео очень низкого качества, но с тех пор они внедрили «пути обучения» и значительно улучшили качество своего контента.
Еще нужно улучшить, но в целом все отлично.
RussianPod101 также учит использовать формат подкастов (обычно два хоста болтают на определенную тему). Также есть видеоролики разной тематики на разных уровнях.
Как и в Rocket Russian, стенограммы и PDF-файлы доступны для скачивания и по большей части имеют высокое качество.
См. Мой обзор RussianPod101.
Что мне нравится:
Достаточно недорого.
Множество уроков русского языка для работы — недостатка в содержании нет.
Что мне не нравится:
Низкое качество, старые видео в некоторых уроках.
Неуклюжий интерфейс.
УНИКАЛЬНОЕ ПРЕДЛОЖЕНИЕ: используйте код MEZZOGUILD , чтобы сэкономить 25% на любом из вариантов курса русского языка.
3. Глоссика русская
Стоимость: 30 долларов в месяц.
Краткое описание: Glossika — один из самых уникальных (но простых) доступных языковых продуктов и, на мой взгляд, один из немногих, в котором используется естественный, основанный на исследованиях метод.Фактически, метод Glossika очень хорошо согласуется с тем, как я лично изучаю языки (включая русский), и я добился огромных успехов в этом.
Я использовал Glossika Russian, когда жил в России, и это помогло мне научиться говорить свободно за несколько месяцев.
Смысл Glossika Russian в частом повторении лексических фрагментов — другими словами, в многократном прослушивании последовательности предложений с естественной скоростью и их повторении. Это, несомненно, самый эффективный тренажер для понимания на слух, который я когда-либо видел.
Грамматика русского языка усваивается естественным путем, без утомительного запоминания правил. Просто много слушал и повторял!
Что мне в нем нравится:
Один из продуктов курсов русского языка, наиболее близких к моему собственному разработанному и проверенному стилю обучения.
Запрещается запоминание грамматики или неестественное изучение языка.
Подходит для изучающих русский язык, которые предпочитают аудио, а не визуальные эффекты.
Доступ ко всем остальным языкам и языковым парам по одной подписке.
Что мне не нравится:
Требуется лучший способ научить студентов тому, как работает этот подход.
Звук на некоторых языках (кроме русского) непостоянного качества.
Сообщил об ошибках в скриптах (лично мне еще не приходилось сталкиваться).
4. Pimsleur Russian
Стоимость: 14,95 долларов в месяц подписки (или 119,95 долларов за уровень).
Краткое описание: Большинство людей слышали о Pimsleur.Это классический фаворит для изучения русского языка с использованием интервалов повторения. Уроки сосредоточены на практической лексике и выражениях, которые могут понадобиться в различных сценариях. Сюда входят приветствия, общие фразы и словарный запас, которые могут вам понадобиться при посещении русскоязычной страны.
Метод Pimsleur подготовит вас к тому русскому языку, который вам необходим в поездках за короткий период времени. Это один из лучших онлайн-курсов русского языка, несмотря на свой возраст, и это доступная программа, на которую можно подписаться или купить полную программу, включающую 30 уроков, за 119 долларов.95.
Прочтите мой обзор Pimsleur.
Что мне нравится:
Pimsleur — это вневременной шедевр, основанный на серьезных исследованиях в области овладения вторым языком
Курс требует всего 30 минут в день на изучение
Есть много прослушиваний и повторений фраз для легкого запоминания
Что мне не нравится:
Примеры устаревших сценариев
Слишком много английского
5.Грамматика Герой (русский язык)
Стоимость: 197 $
Краткое содержание: Grammar Hero был создан моим другом Олли Ричардсом из «Я научу тебя языку». Это серия, которая охватывает несколько языков, включая русский.
Цель Grammar Hero — использовать повествовательные инструкции для упрощения трудных для понимания грамматических понятий на русском языке. Он тщательно знакомит вас с общими проблемами, с которыми сталкивается большинство изучающих русскую грамматику.
См. Мой обзор Grammar Hero.
Что мне нравится:
Подготовлено моим хорошим другом, который, как я могу подтвердить, обладает высокой квалификацией и опытом в области языковой педагогики
Курс отлично справляется с упрощением грамматических понятий русского языка
Охватывает большую часть основного вопросы, которые возникают у новичков
На основе качественных, специально написанных материалов для чтения
Что мне не нравится:
Завышена цена
На мой взгляд, слишком много грамматики
6.Розеттский камень Русский
Стоимость: От 6,49 долл. США в месяц.
Краткое описание: Rosetta Stone — это, безусловно, самое известное имя в области изучения языков и один из самых популярных курсов для изучения русского языка.
Раньше это был дорогой коробочный продукт, но теперь он предлагает доступную онлайн-подписку.
Сила Rosetta заключается в ее уникальном процессе погружения. Поскольку нет перевода или явной грамматики, вы должны изучать словарный запас, грамматику и разговорные навыки с помощью интуиции.Курс интуитивно способствует изучению русского с помощью простых языковых шаблонов по низкой цене подписки.
См. Мой невероятно популярный обзор Розеттского камня.
Что мне нравится:
Не нужно запоминать длинные правила и слова
Уникальный стиль обучения, использующий вашу интуицию
Легко распознавать языковые шаблоны
Что мне не нравится:
Отталкивает людей, ищущих объяснения по грамматике русского языка
Несоответствующие изображения и визуальные подсказки
7.Манго Языки
Стоимость: 7,99 долларов в месяц
Резюме: Я признаю, что за почти десять лет, что я веду этот сайт и просматриваю языковые продукты, я только недавно попробовал Mango Languages.
Хотелось бы попробовать раньше.
Mango Languages реализовала то, что я считаю одним из наиболее интуитивно понятных подходов к разделению на части в своем стиле курса (очень близком к моему личному методу). Он делает это, избегая грамматических объяснений и вместо этого выделяя лексические фрагменты цветом, чтобы помочь вам изучить языковые шаблоны.
Одна из лучших функций, которые я видел в языковом продукте. Период.
Единственная проблема с Mango заключается в том, что он довольно легкий по глубине курса, и я бы хотел, чтобы они пошли дальше для продвинутых изучающих русский и другие языки.
Что мне нравится:
Интуитивно понятный интерфейс «разбиения на части» — одна из лучших реализаций языкового продукта, которые я когда-либо видел. не нравится:
Не хватает глубины курса, который я ожидал бы увидеть на такой замечательной платформе, что делает ее в основном непригодной для учащихся более высокого уровня
8.3 года
Стоимость: Бесплатно (но они, похоже, работают над платным продуктом)
Резюме: 3ears — это драгоценный камень, о котором мало кто говорит. Я с большим нетерпением слежу за их успехами в течение многих лет.
Принцип работы очень похож на FluentU (см. Ниже) — он получает видео с YouTube и предоставляет оболочку с интерактивными элементами субтитров. Вы можете щелкнуть слова в субтитрах, и приложение получит подробную информацию об этом слове (похоже, это машинный перевод).
Очень удобно и (пока) ничего не стоит.
Что мне нравится:
В настоящее время 100% бесплатное использование
Тонны видеоконтента для просмотра и обучения (все с YouTube)
Подробная и исчерпывающая информация о словах, выбранных в интерактивных субтитрах
Что мне не нравится:
Неуклюжий и глючный интерфейс
9. Mondly Russian
Стоимость: От 9 долларов.99 / мес.
Описание: Mondly предлагает курсы для множества разных языков и по стилю похожи на Busuu, Duolingo и Babbel. Даже намеки на розеттский камень в его доставке.
Это красиво оформленное веб-приложение, в котором приятно перемещаться по содержанию курса.
Некоторые курсы не так хороши (например, арабский), но русский и другие проходят очень хорошо.
Что мне нравится:
Отлично подходит для изучения большого словарного запаса
Содержание и упражнения одинаковы для всех уровней
В основном пассивные упражнения
Порядок уроков и тем плохо разработан
Интерфейс неудобен для пользователя и непривлекателен
См. Этот обзор Mondly, чтобы узнать больше.
Другие отличные онлайн-курсы русского языка
Я уже упоминал о своих личных предпочтениях по русскому языку выше, но есть множество других качественных вариантов онлайн-курсов для русского.
Продолжайте читать.
10. Реальный Русский Клуб
Стоимость: Бесплатные + Премиум опции (200 долларов за полную коллекцию)
Резюме: Управляет Дарьей, уроженкой России с огромным количеством подписчиков в Интернете. У нее множество учебных материалов (большинство из них совершенно бесплатно).
Что мне нравится:
Нулевая стоимость бесплатного контента
Видео высокого качества, за которым легко следить
Материал для медленного прослушивания
Что мне не нравится:
Бесплатные материалы урока довольно тонкий — не более того, кроме видео
Обучение в стиле доски не является моим предпочтительным стилем обучения (личные предпочтения)
11. Красная Калинка
Стоимость: 29 € в месяц
Резюме: Красная Калинка — это действительно шведский стол продуктов и курсов русского языка.Сайт существует уже давно и загружен огромным количеством контента.
Контент всеобъемлющий, хотя стиль доставки мне не нравится. Доступ к живому репетитору — определенно большой бонус, хотя его можно легко дополнить другой онлайн-услугой репетитора.
Что мне нравится:
Очень полный и уважаемый
Доступ к личному репетитору включен
Недорого
Что мне не нравится:
Датированный интерфейс
Скучный стиль урока
12.RT (Учите русский)
Стоимость: Бесплатно
Резюме: Вроде как уроки языка BBC, RT предлагает нечто подобное для изучающих русский язык.
Это бесплатный языковой курс, в котором удивительно много деталей, охватывающих множество различных тем. Самое лучшее в нем — это качественный звук.
Я бы сказал, что это удобно в качестве краткого справочника по лексике и выражениям на русском языке, но в ближайшее время вам может потребоваться найти что-то более существенное.
Что мне нравится:
Бесплатно
Вполне всесторонне, учитывая его возраст и нулевую стоимость
Охватывает широкий круг тем, которые полезны для всех изучающих русский язык
Включает аудио
Что я не например:
Серьезно устаревший веб-сайт и интерфейс
Лучше использовать в качестве краткого справочника, чем «курс»
13. Rapid Russian (Earworms MBT)
Стоимость: 13 $.50 за уровень
Резюме: Earworms MBT — один из самых недооцененных и уникальных российских продуктов, которые я когда-либо видел. Он основан на фактических научных исследованиях «ушных червей» (синдром залипшей песни) и их использовании для запоминания языка.
Я добился личного успеха с этим продуктом.
Это был первый продукт, которым я пользовался в России, и это положило начало моему стремлению к изучению языка. Запоминающуюся музыку было легко слушать каждое утро, и она оставалась в моей голове весь день.
Что мне нравится:
Один из самых уникальных и недооцененных продуктов, которые я когда-либо видел и основанный на реальных научных исследованиях
Чрезвычайно эффективный и заставляющий вас запоминать выражения на русском языке
Недорого
Что Мне не нравится:
Требуется выйти за пределы туристического языка
14. FluentU Русский
Стоимость: FluentU стоит 30 долларов в месяц.Пользователи могут получить 4 месяца бесплатно при подписке на полный год по цене 240 долларов США
Резюме: FluentU помогает студентам изучать русский язык с помощью реального видеоконтента. Концепция этого стиля обучения заключается в предоставлении иммерсивного онлайн-курса (своего рода).
Студенты учатся, просматривая сцены из русских видеороликов, которые относятся к реальной русской культуре и содержат русские и английские интерактивные субтитры.
См. Также этот обзор FluentU.
Что мне нравится:
Это отличный вариант для учащихся, которым не нравится много читать.
Доступна двухнедельная бесплатная пробная версия.
Что мне не нравится:
Обратной стороной является то, что он не предлагает никаких интерактивных инструкций, а среда обучения очень нетрадиционна (даже по сравнению с типичным онлайн-курсом), что может быть не идеальным для все учащиеся.
15. Путеводитель по русскому языку
Стоимость: Бесплатно
Резюме: Справочник по русскому языку — это курс в форме блога, который охватывает множество тем, связанных с русским языком и культурой.
Аудио, к сожалению, нет, но это удобный ориентир для изучения русской грамматики и глаголов.
Что мне нравится:
Удобный и краткий справочник по русской грамматике и глаголам
Охватывает довольно много
Бесплатно
Что мне не нравится:
В основном просто блог
В настоящее время нет звука.
16. Колоды (многие курсы русского языка)
Стоимость: Бесплатно
Резюме: По какой-то странной причине Memrise решила, что недавно было бы хорошим маркетинговым решением переместить свое бесплатное «сообщество» курсы на сайт под названием Decks , при наличии премиальной подписки на исходном сайте Memrise.
Насколько я понимаю, колоды идентичны тем, что предлагает Memrise.
Это 100% бесплатные курсы, добавленные сообществом в виде игровой колоды карточек. Вы выбираете язык или диалект, а затем играете в карточную игру «Полив растений». Это вызывает сильное привыкание и на самом деле довольно эффективно.
Не все курсы русского хороши. Ищите те, которые включают аудио и, на мой взгляд, обучают фразам, а не словам.
Что мне нравится:
Повторение облегчает изучение слов
Больше удовольствия, чем традиционные приложения с карточками
Множество курсов на выбор
Что мне не нравится:
Должно быть используется с другими ресурсами или обновляется до платной версии
Не основной инструмент для изучения русского языка
17.Прозрачный Русский
Стоимость: Цены сильно различаются в зависимости от варианта курса
Резюме: Один из самых удивительных онлайн-курсов русского языка, которые я пробовал.
Система и интерфейс довольно устаревшие и медленные, что является реальным недостатком, но если вы можете не обращать внимания на это, Transparent Language обеспечивает реальную глубину русского контента.
Сравнение распознавания голоса в прозрачном языке отсутствует. Он основан на записи вашего голоса и демонстрации вашей звуковой волны для сравнения со звуковой волной носителя языка.
Нет встроенной системы для автоматического сравнения звуков.
Программы, подобные Rocket Russian, раньше делали то же самое, но недавно добавили Google Web Speech для сравнения произношения. Я не знаю, почему Transparent Language не сделал этого, ведь его так легко реализовать.
В курсе «Прозрачный язык» есть предмет «Произведите это. Скажи это.» раздел, который буквально спрашивает вас: «Вы были правы?».
Другими словами, невозможно автоматически определить, были ли вы правы или нет — это зависит от вашего собственного определения.Это невероятно устарело.
В целом, если вы можете не обращать внимания на устаревший дизайн и несовершенный аспект записи голоса, Transparent Language Russian — отличный вариант курса.
Что мне нравится:
Русский диалог на 100% естественная скорость
Обширный охват и глубина содержания
Что мне не нравится:
Устаревший и медленный интерфейс, в котором сложно ориентироваться
В разделе произношения нет встроенного распознавания голоса для сравнения с родным диалогом
18.LingQ
Стоимость: 12,99 $ в месяц
Резюме: LingQ работает уже много лет и был первым продуктом для вспомогательного чтения, который включает русский язык. С тех пор были и другие побочные продукты, которые не так хороши.
Библиотека материалов для чтения и пополнения словарного запаса обширна — недостатка в материалах на русском языке нет.
Мне нравится LingQ, но я лично использую LWT (Learning With Texts), который на 100% бесплатный.
Что мне нравится:
Доступно
Обширная библиотека материалов для чтения
Очень простой в использовании интерфейс
Что мне не нравится:
Ограничено литературным стилем обучения
LWT — бесплатная и лучшая альтернатива, на мой взгляд (но слишком сложная для установки для большинства людей)
19. Мимический метод Русский
Стоимость: Мимический метод стоит 197 долларов и предоставляет пользователям пожизненный доступ к своим материалы.
Краткое описание: Mimic рекламирует курс, полностью посвященный произношению (обращение с языком как с песней). Программа может быть сложной и сложной для понимания, но если ваша единственная цель — улучшить свое произношение, она может быть вам полезна.
Эта программа стоит 197 долларов за штуку.
Некоторую информацию из этого курса можно найти в Интернете бесплатно. В нем легко ориентироваться и он хорошо организован, но все же довольно дорого. Один из самых уникальных российских методов, имеющий фантастические отзывы.
Что мне нравится:
Некоторые домашние задания интересны и уникальны для языковых курсов
В них подробно рассказывается о звуках стихий
Это дает другой способ научиться говорить на языке
Что Мне не нравится:
Слишком дорого
Не так много материала по цене
Не очень интересен и не помогает поддерживать интерес
Требуется пополнение другими ресурсами
Онлайн-курсы русского языка, которые находятся в конце моего списка и не рекомендуются лично
Некоторые онлайн-курсы русского языка популярны, но, на мой взгляд, не так хороши (с личной точки зрения).
Я перечислил их все здесь. Если вы не согласны, оставьте комментарий ниже.
20. Баббель Русский
Стоимость: Babbel имеет хорошие цены на предлагаемые услуги, текущие подписки стоят 12,95 долларов в месяц.
Описание: Еще один вариант одного из лучших онлайн-курсов русского языка, Babbel, является отличным инструментом для изучения языка и очень экономичен. Платформа онлайн-курсов русского Babbel ориентирована на письмо и чтение, аудирование и правописание.Его цель — дать вам все инструменты, необходимые для немедленного начала изучения языка.
Babbel доступен для предприятий, которым необходимо, чтобы их сотрудники изучали русский язык. Он также доступен в качестве подарка. Благодаря множеству доступных пакетов легко найти тот, который подходит для ваших потребностей в изучении русского языка. Babbel взимает ежемесячную плату, а пакеты варьируются от 6 до 12 долларов в месяц.
Считайте это платной и немного лучшей версией Duolingo.
Прочтите этот обширный обзор Babbel.
Что мне нравится:
Гибкость и разнообразие стилей преподавания
Включает несколько разных стилей обучения для эффективного преподавания русского языка
Позволяет самостоятельно изучать язык
Что мне не нравится:
Никто не говорит с родным акцентом
Нет материалов для скачивания
Слабые разговорные навыки
21. Duolingo Русский
Стоимость: Бесплатно.
Краткое описание: Duolingo теперь стало нарицательным и используется школой в качестве дополнительной программы на своих языковых курсах. Это весело и бесплатно, хотя у него есть платное обновление подписки, которое удаляет добавлений и дает дополнительные ресурсы за несколько долларов в месяц.
Идея Duolingo состоит в том, чтобы сделать изучение русского увлекательным и побудить студентов возвращаться к ним каждый день благодаря игровому обучению. Русский язык изучается с помощью различных упражнений и большого количества повторений.
Лично не фанат, но это бесплатно.
Посмотрите это сравнение Duolingo и Rosetta Stone.
Что мне нравится:
Интерактивные уроки позволяют говорить и слушать на русском языке
Предлагает соревнования между друзьями и взаимодействие с другими пользователями
Отлично подходит для знакомства с языком и изучения основ
Что мне не нравится:
Аудиоголос может звучать немного неестественно
Не совсем всесторонне, чтобы говорить свободно
22.Мишель Томас Русский
Стоимость: Начинается с $ 11,99
Резюме: Некоторое время назад я подробно рассмотрел Мишеля Томаса и в итоге почти ничего не сказал о курсе (оба уровня). Я получил горы критики за отзыв! На мой взгляд, метод Мишеля Томаса полон дыр и бросает вызов текущим выводам исследований в SLA.
Основная посылка Мишеля Томаса заключается в том, что вы расслабляетесь, как клиент в кресле психолога, и позволяете учителю контролировать ваше обучение.Вам говорят: «никогда не пытайтесь учиться», без запоминания, без практики, без ответственности ученика.
Учителя Мишеля Томаса помогают ученикам исправлять каждую ошибку на месте .
Во время занятий нет тренинга на восприятие речи на слух и нет возможности для естественного разговора.
В целом, русский Мишель Томас может быть полезным учебником, но лично я бы никогда не рекомендовал этот курс.
Прочтите мой подробный и весьма противоречивый обзор Мишеля Томаса для получения дополнительной информации.
Что мне нравится:
Нет необходимости писать или запоминать
Грамматика вводится естественным образом и постепенно
Что мне не нравится:
Под руководством учителя.
Постоянное исправление ошибок.
Не готовит русских студентов к разговору и восприятию на слух.
23. Busuu русский язык
Стоимость: От 5,83 доллара в месяц.
Резюме: Busuu — это еще одна программа, похожая на Babbel и Duolingo, однако она не так структурирована, как другие курсы, и содержит меньше инструкций по словарному запасу до начала важных компонентов.
В целом дизайн Busuu красив, но ему просто не хватает глубины.
Существует бесплатная версия Busuu с разделом языкового обмена. Это хороший выбор. Однако их служба подписки, которая стоит более 8 долларов в месяц при минимальном трехмесячном сроке, может быть не лучшим соотношением цены и качества.
Что мне нравится:
Учебная платформа хорошо спроектирована
Языковой обмен — бесплатное, простое в использовании приложение
Очень популярная программа онлайн-обучения
Что мне не нравится:
Программные уроки оставляют желать лучшего
Недостаточно произношения и грамматики
Лучшие курсы доступны бесплатно
См. Этот обзор Busuu.
24. ФГИ России
Стоимость: Бесплатно
Резюме: FSI (Институт дипломатической службы) — государственное учреждение, которое обучает дипломатов и государственных служащих иностранным языкам.В настоящее время в Интернете можно бесплатно загрузить курс русского языка (включая аудио).
Проблема с материалом FSI в том, что он существует буквально почти столетие.
Старый. Действительно старый.
Итак, хотя вы можете бесплатно скачать исчерпывающий подробный курс русского языка со звуком, имейте в виду, что материал представляет собой буквально фотокопии буклетов, напечатанных на пишущих машинках, что делает его использование невыносимым.
Если вы терпеливы, курсы FSI имеют некоторую ценность, но они настолько устарели, что я лично не стал бы беспокоиться.
Что мне нравится:
Будучи государственной организацией США, которая занимается обучением дипломатов, FSI, естественно, имеет невероятную глубину курса.
Бесплатно и легко скачать урок + аудио на многих сайтах.
Что мне не нравится:
Очень старый курс.
PDF-материал — это всего лишь фотокопия оригинальной машинописной бумаги, поэтому читать его ужасно.
25. Live Lingua
Стоимость: Начинается с 10 долларов.99 в час.
Краткое описание: Live Lingua подберет для вас частного репетитора русского языка, говорящего на родном языке, и позволит вам учиться в удобном для вас темпе. Вы можете пройти пробный урок бесплатно.
Хотя их уроки доступны для частного репетитора, начиная с 16 долларов в час, вы можете получить скидку на комплект. По той цене, которую они взимают, вы можете найти более экономичным просто использовать italki .
Что мне нравится:
Частные уроки один на один по Skype
Предоставляет широкий спектр материалов для изучения русского языка
Поддержка многих программ
Что мне не нравится:
Дороже, чем другие онлайн-репетиторы русского языка
Варианты не такие гибкие, как другие программы
Использует бесплатные курсы с других сайтов
26.Живой язык
Стоимость: Начинается с 10,99 долларов в час.
Резюме: Я никогда не был поклонником Living Language с тех пор, как просмотрел его несколько лет назад. Я включаю его в этот список, потому что это один из самых громких и популярных курсов для таких языков, как русский (к тому же меня время от времени спрашивают об этом).
Я лично считаю Живой язык мягким, неправильно выровненным и просто очень скучным курсом с тяжелой грамматикой.
Определенно не моя чашка чая (хотя может подойти людям, любящим грамматику).
Что мне нравится:
Достаточно подробные объяснения по русской грамматике
Что мне не нравится:
Неправильное выравнивание — особенно для более высоких уровней
Убийство конкурентов (например, Розеттский камень) в своем маркетинге очень отталкивает
Резюме: Лучшие онлайн-курсы русского
Я думаю, что я рассмотрел практически все доступные онлайн-курсы русского языка в настоящее время (дайте мне знать в разделе комментариев, если я пропустил один) .
Независимо от того, какой курс русского языка вы выберете, частая практика русского языка с носителями языка является обязательной; в идеале лично, если можете (еще лучше, если вы можете сделать это за границей).
Если найти рядом с вами носителей языка невозможно, то italki — отличная платформа для поиска действительно доступных русских партнеров и репетиторов.
В целом ваш успех определяется вашей решимостью и исходной мотивацией.
Даже если вы пройдете все курсы из этого списка, вы все равно не сможете хорошо освоить русский язык без должной мотивации и последовательности.
Точно так же даже менее выдающиеся курсы русского языка могут иметь большое значение в руках человека с правильным отношением и стилем обучения.
Если вы ищете личные советы о том, как выучить русский язык и преодолеть различные трудности в изучении языка (на основе моих собственных исследований и опыта), обязательно подпишитесь ниже, выбрав «Вступление в гильдию».
Знаете курс русского языка, который я не упомянул?
Поделитесь им в комментариях.
6 лучших онлайн-инструментов для проверки грамматики для WordPress (2021)
Вы ищете лучшие онлайн-инструменты для проверки грамматики для WordPress? Большинство браузеров также имеют встроенную проверку орфографии, но она не проверяет грамматику и удобочитаемость. В этой статье мы выбрали лучшие инструменты проверки грамматики, которые вы можете использовать с WordPress.
Зачем добавлять инструмент проверки грамматики для WordPress?
Ошибаются даже самые опытные писатели.Вычитка собственных статей часто приводит к небольшим ошибкам по неосторожности, потому что мы часто торопимся с контрольным списком перед публикацией.
Один из способов решить эту проблему — прочитать свои статьи вслух. Слушание собственного голоса может помочь вам найти ошибки и улучшить общую оценку читаемости ваших статей.
Чтобы помочь вам с грамматикой и орфографией, вы можете использовать инструмент проверки грамматики. Эти инструменты могут обнаруживать орфографические и грамматические ошибки при вводе текста. Это экономит ваше время на корректуру и улучшает содержание вашего сайта.
При этом давайте взглянем на лучшие инструменты проверки грамматики для WordPress, которые вы можете использовать.
1. Грамматика
Grammarly — это популярный онлайн-инструмент для проверки грамматики, доступный в качестве надстройки браузера для Google Chrome, Firefox и даже Microsoft Edge. Он проверяет грамматические и орфографические ошибки, когда вы пишете свои сообщения.
В правом нижнем углу области ввода появится индикатор. Щелкнув по индикатору, вы увидите количество ошибок.Орфографические, грамматические и контекстные ошибки будут подчеркнуты во время написания.
Одним из недостатков Grammarly является то, что вам придется переключиться на текстовый редактор для корректуры.
2. Имбирь
Ginger — еще один удобный инструмент для проверки орфографии и грамматики, который поможет улучшить ваши навыки письма и повысить производительность. Он доступен как надстройка для браузера и предлагает приятный пользовательский интерфейс.
Он отображает крошечный значок в правом нижнем углу областей ввода, включая редактор сообщений WordPress, Gmail, Facebook и многое другое.Он проверяет ошибки при вводе и выделяет ошибки в контексте.
У использования имбиря есть несколько недостатков. Вам нужно будет переключиться на текстовый редактор в WordPress. Для лучшей вычитки вам нужно будет щелкнуть значок, чтобы запустить редактор Ginger.
3. По истечении срока
After The Deadline доступен как расширение Google Chrome. Он поставляется со встроенным инструментом проверки орфографии с дополнительной проверкой грамматики и удобочитаемости.
После того, как вы установили надстройку в своем браузере, вам нужно будет щелкнуть по ней и выбрать параметры.Это откроет страницу настроек надстройки, где вы можете проверить параметры, которые вы хотите, чтобы надстройка обращала внимание.
Вы можете запустить инструмент во время написания сообщений в блоге, щелкнув значок проверки орфографии в правом нижнем углу редактора.
4. Хемингуэй
Hemingway доступен в виде текстового веб-редактора, поэтому он не работает непосредственно в редакторе сообщений WordPress. Однако вы можете использовать их бесплатный веб-редактор, чтобы проверить свои статьи на удобочитаемость, грамматику и орфографические ошибки.
Думайте о Хемингуэе не только как о грамматике, но и как о проверке стиля. Он разработан для улучшения читабельности, показывая вам оценку читабельности вашего контента и предлагая улучшения. Веб-приложение имеет чистый макет, который не отвлекает внимание, который может помочь вам быстро оценить контент, прежде чем нажимать кнопку публикации в WordPress.
Hemingway также доступен как платное настольное приложение для Windows и Mac, которое позволяет публиковать сообщения непосредственно в WordPress.
5. LanguageTool
LanguageTool доступен как бесплатное дополнение для Chrome, FireFox, Google Docs и LibreOffice. Он также доступен в виде веб-редактора и полноценного настольного приложения.
Преимущество LanguageTool в том, что он работает с английским, немецким, польским, русским и более чем 20 другими языками. Это делает его особенно полезным инструментом для многоязычных веб-сайтов и многоязычных авторов.
Обратной стороной является то, что бесплатная версия ограничена только 20 000 символов на проверку, поэтому вам нужно будет перейти на платные планы, чтобы получить больше функций.
6. Реактивный ранец
Jetpack — это набор плагинов для WordPress, который содержит множество полезных модулей. Одна из его основных функций — добавление корректуры в редактор сообщений WordPress.
Для расширенной проверки грамматики вам необходимо посетить страницу JetPack »Настройки . На вкладке письма вы увидите раздел орфографии, стиля и грамматики. Отсюда вы можете включить различные параметры корректуры, включая правила стиля.
Обратной стороной использования JetPack является то, что для этого требуется WordPress.com, и вы также получите множество функций, которые могут вам не понадобиться.
Мы надеемся, что эта статья помогла вам найти лучшие онлайн-инструменты проверки грамматики для WordPress. Вы также можете увидеть наш список лучших плагинов для контактных форм для WordPress.
Если вам понравилась эта статья, то подпишитесь на наш канал YouTube для видеоуроков по WordPress. Вы также можете найти нас в Twitter и Facebook.
Какой онлайн-переводчик выбрать?
Перевод текста с русского на английский с помощью присяжного профессионального переводчика будет стоить вам в среднем 0 долларов.10 слов. При устном переводе переводчиками считайте около 400 долларов в день …
Так почему бы не научиться переводить в одиночку?
Услуги перевода, безусловно, будут лучше, но с небольшой работой и помощью, благодаря программному обеспечению для перевода, вы можете стать асом в переводе с русского на английский. Еще одна причина для изучения русского — ознакомьтесь с нашими советами по онлайн-курсам русского языка.
Вот сравнение различных онлайн-переводчиков и советы по профессиональному переводу.
Как выбрать лучший онлайн-переводчик с русского на английский?
Онлайн-переводчики значительно улучшились за последние годы. И если преподаватели языка посоветуют или запретят их использование, они дадут вам точное представление о содержании текста.
Очевидно, что для перевода текста требуется знание языка. Таким образом, мы не советуем вам пытаться переводить текст с английского на русский, не зная языка. Результат может быть смешан с неверными толкованиями.
Качество онлайн-переводчиков двойное или низкое. Некоторые из них просто переводят слово в слово, создавая предложения с сомнительным синтаксисом, в то время как другие делают глобальный анализ текста.
Чтобы выбрать лучшего онлайн-переводчика , учитывайте два критерия :
, насколько легко им пользоваться: понятно, без рекламы и быстро.
Качество перевода: с учетом орфографии, идиоматических выражений, синтаксиса, фразовых глаголов, имен собственных и т. Д.
Если большинство инструментов онлайн-перевода набирают от 50 до 60% успешного перевода представленных текстов, то именно Google Translation получает все награды с 80% (для большинства языков).
Тем не менее, если переводчики позволяют шлифовать работу, они не заменят человека для исправления ошибок и ретуширования иногда немного шатких предложений — по крайней мере, пока!
Подумайте о русских видео, чтобы прогрессировать в словарном запасе.
Google Translate: мировой лидер
Кириллица усложняет перевод для английского студента.
Лучший англо-русский переводчик — это, без сомнения, Google Translate. Чистый стиль, простой в использовании, ясный, но, прежде всего, лучший перевод с точки зрения перевода.
Не будучи профессиональным переводчиком, Google Translate полагается на колоссальную базу данных и скрещивает тысячи переводов , чтобы предоставить вам лучшее.
Кроме того, Google Translate предлагает вам оставлять отзывы для постоянного улучшения. Инструмент также является интерактивным и дополняется комментариями каждого пользователя Интернета.
Некоторые функции очень полезны, особенно во время путешествий по России, например:
Перевод с фотографии / изображения: просто выберите язык, на который вы хотите перевести текст, выделите этот текст, а Google позаботится обо всем остальном. ,
Загрузите приложение Google Translate и используйте его для мгновенного автономного перевода,
Практикуйте произношение с помощью голосового интеллекта инструмента.
Более того, он интегрирован непосредственно в браузер Google Chrome, что позволяет при необходимости переводить веб-сайт на ваш родной язык.
Не забудьте читать книги на русском языке чтобы пополнить словарный запас.
Русско английский переводчик: Reverso, лучший с точки зрения идиоматических выражений
Перевод на русский язык — это еще и встречные выражения, как в английском!
Reverso также имеет очень хорошие характеристики и даже превосходит Google Translate по переводу идиоматических выражений.
Таким образом, Reverso будет правильно переводить «Мне по барабану» как «Мне все равно», в то время как Google Translate дословно переведет «меня по барабану» …
С другой стороны, мы вернемся к дизайн сайта, достойный 1990-х годов, и реклама, занимающая большую часть окна. Но перевод хороший и тем более бесплатный, в отличие от бюро переводов.
Здесь тоже сайт коллаборативный. Каждый пользователь может оставить свое мнение о переводе текста, хороший или плохой, и может добавить слова из лексики.
Reverso также является словарем, средством проверки орфографии, проверки спряжения и средством проверки грамматики русского языка. Это позволяет избежать очевидных ошибок, которые обычно допускают начинающие переводчики.
Кроме того, если в вашем переводе существует несколько возможностей, Reverso покажет вам лучший вариант в соответствии с контекстом .
Многие сайты позволяют изучать русский язык.
Русско-английский переводчик Вавилон: для перевода в специализированной области
Вавилон поможет вам выучить русский язык и, в частности, обогатить ваш словарный запас и знание русских слов в соответствии с контекстом.Таким образом, инструмент предлагает бесплатный перевод конкретных терминов, относящихся к конкретным областям:
Технический перевод,
Юридический перевод,
Медицинский перевод,
Маркетинговый перевод,
Коммерческий перевод технической документации и т. Д.
Онлайн-переводчик Babylon эффективен, поскольку он учитывает контекст текста, отправляемого на перевод, чтобы предложить вам лучшее.
Подкасты на русском языке могут обогатить ваши знания языка.
Онлайн-словари
Русская клавиатура часто доступна онлайн.
Автоматический перевод онлайн-переводчиков показывает некоторые недостатки на языке Достоевского. Присутствие человека необходимо, и для перевода русского текста недостаточно знать русский алфавит и сделать дословный перевод.
Англо-русский словарь настоятельно рекомендуется тем, кто занимается переводческим искусством при подготовке к поселению в Санкт-Петербурге или при выборе профессии русского переводчика.
Если Google Translate, Reverso и Babylon могут предоставить вам некоторые правильные элементы на славянских языках, то эй не заменит качество и полноту русско-английского словаря.
Давайте остановимся на онлайн-словарях, чтобы продолжить дематериализацию. Кроме того, он по-прежнему занимает гораздо меньше места, чем словарь в его физической версии!
Лучшее в своей категории, будь то перевод на английский, испанский французский или немецкий французский, это, несомненно, WordReference .Он доступен только в русско-английском или русско-английском переводе.
Главное достоинство этого словаря — очень хороший перевод в контексте . Таким образом, если слово имеет несколько значений, всем им будут даны контекстные, а иногда и выражения, в которых это слово может использоваться.
Если вы не можете найти свое счастье (такое может случиться) среди ответов, вы также можете воспользоваться форумом, чтобы задать свои вопросы или найти ответ.
Для дословного перевода с русского на английский переведите на dict.com . Здесь снова перевод слова детализирован в зависимости от его использования в разных контекстах. Как и в примере ниже, русское слово «пить» также означает «пила».
Русско-английский словарь незаменим!
Бесплатный онлайн-перевод требует проверки человеком.
6 шагов, которые необходимо выполнить для хорошего русско-английского перевода
Помимо прохождения курса русского языка с русским учителем, чтобы больше узнать о русской культуре, прежде чем приступить к русскому переводу с английского с головы вниз, необходимо выполнить несколько шагов.
Прочтите несколько раз исходный текст на исходном языке. Даже если вы не понимаете всего этого, это хороший способ понять общую идею.
Используйте метод воронки: от самого широкого к самому точному. Начните с предварительного перевода, сохраняя общий тон текста, а затем настраивайте его по мере чтения,
Отметьте или выделите неизвестные выражения. С контекстом вы можете не закончить их понимание,
Избегайте анахронизмов,
Используйте онлайн-словарь для самых сложных слов, если вы действительно не понимаете значения,
Правильно прочитайте его перевод, чтобы выявить ошибки спряжения, грамматики и синтаксис во время уроков русского языка.
Поначалу автоматический переводчик действительно может вам помочь. Но для тренировок и прогресса ничто не сравнится с работой в одиночку с минимальным использованием этих замечательных онлайн-инструментов.
Русско-английский перевод: ошибок, которых следует избегать!
Для присяжного перевода ничто не сравнится с вмешательством человека.
Русский текстовый процессор допускает несколько типичных ошибок. Вот подводные камни, которых следует избегать:
Дословный перевод: само собой разумеется, что русский язык сложен и имеет свои собственные выражения, которые не будут правильно переданы при дословном переводе,
Перевести или недооценить: в английском тексте не должно быть больше или меньше, чем в исходном тексте.Вы должны быть верны исходному тексту. Художественный перевод должен вызывать более устойчивый тон, чем перевод газетной статьи, например,
Совершите ерунду: напишите фразу или предложение, которые абсолютно ничего не значат. Перечитайте себя, чтобы избежать этого,
Сделайте ложный смысл: то есть путайте одно слово с другим.
Сделать неверное толкование: то есть перевести слово, противоположное истинному значению. Это может очень раздражать в юридическом или медицинском контексте, например,
Пропуск: забыть все или часть предложения.Это может радикально изменить смысл предложения,
Орфографические, временные или синтаксические ошибки.
Чтобы быстрее развиваться на русском языке с русскоязычным приложением на вашем смартфоне.
Sumeree:
Google Translation и Reverso — очень хорошие инструменты для перевода с русского на английский.
Но ничто не заменит человеческую проверку, чтобы гарантировать качество перевода.
С помощью русско-английского словаря при некотором знании языка можно получить квазипрофессиональный перевод .При условии, что вы не набрасываете какую-то странную чушь, суб-перевод или неверную интерпретацию на уроках русского для начинающих.
Если вы хотите найти репетитора, поиск курсов русского языка в Лондоне дает больше всего результатов на Superprof, но если вы изучаете русский язык онлайн, выбор гораздо больше.
Генератор фальшивого русского акцента — Говори / пиши
Поиск инструмента
Русский акцент
Инструмент для имитации русского акцента.Чтобы придать русскому акценту достоверность, текст должен воссоздавать особенности советского акцента.
Результаты
Русский акцент — dCode
Тег (и): Развлечения / Разное, Система связи
Поделиться
dCode и другие
dCode является бесплатным, а его инструменты являются ценным подспорьем в играх, математике, геокешинге, головоломках и задачах, которые нужно решать каждый день! Предложение? обратная связь? Жук ? идея ? Запись в dCode !
Рекламные объявления
Генератор русского акцента
Текст для переписывания с русскоязычным произношением dCode расшифровывает русский язык, это мой товарищ. Язык текста EnglishFrench (Français) Имитация русского акцента
Ответы на вопросы (FAQ)
Как говорить с русским акцентом?
Чтобы знать, как написать или произнести русский акцент , имитатор должен проанализировать его: русский акцент может быть получен, если взять более низкий тон и адаптировать произношение, например, вращение r.
Неизбежные в каждом фильме про Холодную войну, СССР, Москву или русскую мафию, русские персонажи всегда с суровым и неприятным голосом.Советская фонология имеет произношение с жесткими звуками. Сторонники Советского Союза также подвергаются стигматизации, называя всех товарищами.
Пример: С помощью этого переводчика акцента dCode на русский язык переводится dKode на Rrrussian
Если возможно, удалите такие статьи, как a или. И прежде всего сказать «да» с Да!
Имитация русского языка частично схожа с немецким акцентом.
У русских с востока России несколько другие акценты и диалекты, возможно, из-за их близости к Китаю.
Как накатить R как русский?
Прокрутка / трель буквы R для русского акцента , техника аналогична во многих языках (испанский акцент, итальянский акцент, португальский акцент, английский акцент и т. Д.).
R Звук создается за счет вибрации языка на тыльной стороне верхних зубов.
Как слушать голос или звук?
dCode не предлагает аудио преобразование русского акцента , а дает представление о его русском произношении в письменной форме.Чтобы прослушать русский текст, используйте Google Translate: здесь (ссылка)
Задайте новый вопрос
Исходный код
dCode сохраняет за собой право собственности на исходный код онлайн-инструмента «Русский акцент». За исключением явной лицензии с открытым исходным кодом (обозначенной CC / Creative Commons / бесплатно), любого алгоритма, апплета или фрагмента с «русским акцентом» (конвертер, решатель, шифрование / дешифрование, кодирование / декодирование, шифрование / дешифрование, переводчик) или любого «русского акцента» ‘функция (вычислить, преобразовать, решить, расшифровать / зашифровать, расшифровать / зашифровать, декодировать / закодировать, перевести), написанная на любом информационном языке (Python, Java, PHP, C #, Javascript, Matlab и т. д.)), и никакая загрузка данных, скрипт, копипаст или доступ к API для «Русского акцента» не будут бесплатными, то же самое для автономного использования на ПК, планшете, iPhone или Android! dCode распространяется бесплатно и онлайн.
Нужна помощь?
Пожалуйста, посетите наше сообщество dCode Discord для получения помощи! NB: для зашифрованных сообщений проверьте наш автоматический идентификатор шифра!
Вопросы / комментарии
Сводка
Похожие страницы
Поддержка
Форум / Справка
Ключевые слова
русский, акцент, россия, ссср, советский, советский, произношение, фонология, диалект, язык, москва, мимика, имитация, английский, речь
Некоторые ресурсы для вычислительной работы по русской морфологии и фонологии
россиян все оцифровывают и выкладывают в интернет. Это упрощает работу корпуса над языком.
Вероятно, наиболее полезным стартовым ресурсом для тех, кто интересуется русской морфофонологией, является классический словарь Зализняка 1977 года ( Грамматический словарь русского языка) , Москва: Русский язык.Он доступен в нескольких форматах в Интернете:
Обратный список форм. Вроде как словарь рифм.
Полный список парадигм Зализняка (TXT в файле RAR): около 90 000 флективных форм русских слов с ударением. Автоматически генерируется из словаря Зализняка Андрея Усачева. Содержит несколько ошибок (даны некоторые неточные краткие формы прилагательных, а пробелы в парадигмах иногда заполняются неправильно), но в остальном весьма полезен.
Интернет-версия базы данных
: введите слово, и эта база данных вернет все грамматические коды оригинала 1977 года, включая тип ударения, класс склонения и т. Д.
Версия для скачивания. Это файл Windows .exe, но вы можете извлечь файл .dbf, который содержит всю информацию в онлайн-версии. После этого файлы DBF можно импортировать в R в любой ОС.
Частота и поиск по корпусу
Ruscorpora: Доступный для поиска веб-корпус русских текстов и устной речи.
Списки частот и лемм Сержа Шароффа; включает такие вещи, как списки двух-, трех- и тетраграммы (орфографические строки различной длины, упорядоченные по частоте встречаемости).Вы, вероятно, не сможете использовать это, если не умеете читать по-русски или хотя бы не владеете кириллицей.
Частотные словари: Russian lg page / English translation Несколько небольших частотных словарей русского языка, включая список частот лемм для 5000 наиболее часто встречающихся слов и некоторую информацию о средней длине слова и так далее.
Яндекс. Доминирующая российская поисковая система. По умолчанию он ищет русские слова во всех падежных формах, поэтому вы получаете приблизительное количество лемм.
Словари разные
Академические словари: онлайн-словари, включая Ожегова, Ефремова, этимологический словарь Фасмера, Даля и многие, многие другие (вы знали, что у филателистов есть свой словарь?).Исчерпывающий, точный и закодированный в UTF-8.
Загружаемые словари: Сборник ссылок на загружаемые словари. Не все являются точными или полными (например, словарь Зализняка не отображается в полной форме, но вы можете восстановить информацию из имеющихся частей).
Словарь заимствований Лёхина и Петрова: онлайн-версия с возможностью поиска.
Rosenthall’s Spelling Reference: все, что вам нужно знать об особенностях русской орфографии и пунктуации.Некоторые полезные обсуждения чередования гласных и согласных, представленных в орфографии, но не в устной речи.
Словарь лингвистической терминологии Ахмановой. На русском.
Другие языки: корпуса и списки слов
На странице проекта CompSeg Learner около двух десятков лексик, подходящих для фонологической работы. Многие из них были подготовлены Джульет Стэнтон или мной, а некоторые — от таких людей, как Майкл Беккер.
An Crúbadán: Корпус для языков меньшинств: коллекция загружаемых орфографических корпусов на сотнях языков, включая тексты из Википедии, переводы Библии, Декларацию прав человека, блоги и твиты.
Списки слов IPA
Кая Шотта [[a-l]] [l-y]: они содержат основанные на орфографии транскрипции IPA для списков слов, которые составляют основу для свободно доступных словарей программы проверки орфографии OpenOffice. Словари были созданы в рамках проекта Саймона Шотта, веб-сайт которого в настоящее время не функционирует. Вам нужно перепроверить транскрипции IPA с описаниями языков, прежде чем использовать их для работы с фонологическим корпусом.
Подготовка лингвистических документов
LyX и LaTeX
Библиография и файлы texmf
Мой каталог texmf со всеми пакетами и стилями LaTeX, которые я использую для лингвистической работы.
Мой файл .bib — в основном это фонология, русский язык и морфология; 4800+ записей. На молнии.
Работа с международным фонетическим алфавитом
IPA_SIL Раскладка клавиатуры IPA для Mac OS, которую я считаю наиболее интуитивно понятной и удобной для пользователя. Файл .zip включает документацию. Этот макет изначально распространялся sil.org, но, похоже, был удален.
Использование шрифтов IPA и раскладок клавиатуры Прагматично, а не исчерпывающе.Если вы хотите узнать все о Unicode или устаревших шрифтах, лучше всего самостоятельно поискать в Интернете.
Деревья
Praat
Введение в основные функции Praat для курса «Звук и язык».
Звуки мира
Небольшая коллекция ссылок и ресурсов для студентов, изучающих фонетику.
Разное
Очень короткий раздаточный материал с регулярными выражениями, который я написал для своего курса казахских полевых методов. Он объясняет основы регулярных выражений для лингвистов с фонологическими примерами.
Как использовать MS Word как профессионал Это очень старый раздаточный материал, но если вы приверженец MS Word, вы все равно можете найти его полезным. В нем описаны некоторые полезные и хорошо скрытые функции Microsoft Word, о которых следует знать лингвистам, такие как полуинтеллектуальная автоматическая последовательная нумерация и перекрестные ссылки.
Как запустить R на более чем одном ядре ЦП в Mac OS с помощью screen Обычно R.app работает только на одном ядре в Mac OS. Этот совет поможет вам выполнять в R ресурсоемкие задачи одновременно, используя Терминал и утилиту screen.
Шпаргалка по Git
Проверка грамматики и орфографии — расширение LanguageTool
★ Находит множество ошибок, которые простая проверка орфографии не может обнаружить ★ Регистрация не требуется ★ Поддерживает более 25 языков (см. Ниже) ★ Работает практически на любом веб-сайте, включая Gmail, Facebook, Twitter
С помощью этого расширения вы можете проверять текст с помощью программы проверки стиля и грамматики LanguageTool. LanguageTool находит множество ошибок, которые простая проверка орфографии не может обнаружить, например, смешивание там / их, a / an или повторение слова, и может обнаруживать некоторые грамматические проблемы.Он поддерживает более 25 языков, включая английский, испанский, французский, немецкий, польский и русский.
LanguageTool разработан с расчетом на простоту. Он мгновенно проверяет орфографию и грамматику любого текста в текущем текстовом поле. LanguageTool совместим практически со всем текстом, независимо от источника, включая социальные сети, такие как Twitter или LinkedIn, и онлайн-сервисы электронной почты, такие как Gmail. Очень немногие сайты, такие как chrome.google.com, в настоящее время не поддерживаются.
С помощью LanguageTool наша цель — предоставить пользователям полнофункциональную программу проверки грамматики и корректора, которая позволит им контролировать свой контент и быть уверенным в этом.LanguageTool является жизненно важным расширением для людей, для которых язык не является родным, но он также достаточно умен, чтобы распознавать многие ошибки, которые обычно допускают носители языка. Это исправление грамматики и орфографии работает во всех вариациях общего языка; например, различая американский и британский английский. LanguageTool также имеет персональный словарь для исключений или слов, которые вы можете часто использовать, но которых нет в обычном словаре. LanguageTool распознает эти слова при будущей проверке грамматики и корректуре.
LanguageTool работает для многих языков — в отличие от Grammarly (также известного как Grammerly :-), Ginger и ProWritingAid.
Ваша конфиденциальность важна для нас: по умолчанию это расширение проверяет ваш текст, отправляя его на https://languagetool.org через надежно зашифрованное соединение. Для использования этого расширения учетная запись не требуется. Мы не храним ваш IP-адрес. См. Https://languagetool.org/privacy/ для ознакомления с нашей политикой конфиденциальности.
Отправляйте сообщения об ошибках или вопросы на https: // forum.Languagetool.org
Список поддерживаемых языков: английский (австралийский, канадский, Великобритания, Новая Зеландия, ЮАР, США), французский, немецкий (Австрия, Германия, Швейцария), астурийский, белорусский, бретонский, каталонский (также валенсийский), китайский, датский, Голландский, эсперанто, галисийский, греческий, итальянский, японский, кхмерский, персидский, польский, португальский (Бразилия, Португалия, Ангола, Мозамбик), румынский, русский, словацкий, словенский, испанский, шведский, тагальский, тамильский, украинский, арабский
С давних времен люди собирали сведения о том мире, в котором они живут. Была лишь одна наука, объединяющая всю информацию о природе, которую человечество накопило на тот момент. Тогда еще люди не знали, что они наблюдают примеры физических явлений. В настоящее время такая наука носит название «естествознание».
Что изучает физическая наука
Со временем научные представления об окружающем мире заметно изменились – их стало гораздо больше. Естествознание раскололось на много отдельных наук, среди которых: биология, химия, астрономия, география и другие. В ряде этих наук не последнее место занимает физика. Открытия и достижения в этой области позволили человечеству обладать новыми знаниями. К ним можно отнести структуру и поведение различных объектов всяких размеров (начиная с гигантских звезд и заканчивая мельчайшими частицами – атомами и молекулами).
Физическое тело — это…
Существует специальный термин «материя», которым в кругах ученых называют все, что есть вокруг нас. Состоящее из материи физическое тело — это какое-либо вещество, занимающее определенное место в пространстве. Любое физическое тело в действии можно назвать примером физического явления. Опираясь на это определение, можно сказать, что любой предмет является физическим телом. Примеры физических тел: кнопка, блокнот, люстра, карниз, Луна, мальчик, облака.
Что такое физическое явление
Любая материя находится в постоянном изменении. Одни тела двигаются, другие соприкасаются с третьими, четвертые крутятся. Не зря много лет назад философом Гераклитом была произнесена фраза «Все течет, все меняется». У ученых есть даже специальный термин таким изменениям – это все явления.
К физическим явлениям относится все то, что движется.
Какие существуют типы физических явлений
Это явления, когда из-за воздействия температуры некоторые тела начинают трансформироваться (изменяется форма, размер и состояние). Пример физических явлений: под воздействием теплого весеннего солнца тают сосульки и превращаются в жидкость, с наступлением холодов лужи замерзают, кипящая вода становится паром.
Механические.
Эти явления характеризуют смену положения одного тела по отношению к остальным. Примеры: часы идут, мяч прыгает, дерево качается, ручка пишет, вода течет. Все они находятся в движении.
Электрические.
Характер этих явлений полностью оправдывает свое название. Слово «электричество» уходит корнями в греческий язык, где «электрон» значит «янтарь». Пример достаточно простой и многим наверняка знакомый. При резком снятии с себя шерстяного свитера слышится небольшой треск. Если проделать это, отключив в комнате свет, то можно увидеть искорки.
Тело, участвующее в явлении, которое связанно со светом, называют светящимся. В качестве примера физических явлений можно привести всем известную звезду нашей Солнечной системы – Солнце, а также любую другую звезду, лампу и даже жучка-светлячка.
Распространение звука, поведение звуковых волн при столкновениях с препятствием, а также иные явления, которые так или иначе связаны со звуком, относятся к этому типу физических явлений.
Они происходят благодаря свету. Так, например, человек и животные способны видеть, потому что есть свет. В эту группу также включены явления распространения и преломления света, его отражение от предметов и прохождение сквозь разные среды.
Теперь вы знаете, какие бывают физические явления. Однако стоит понимать, что между природными и физическими явлениями существует определенная разница. Так, при природном явлении происходит одновременно несколько физических явлений. Например, при ударе молнии в землю происходят следующие явления: магнитное, звуковое, электрическое, тепловое и световое.
fb.ru
Ответы@Mail.Ru: что такое физические явления?
Физическими явлениями называют такие явления, которые обнаруживаются посредством физических действий, как, например, шум, движение и перемещение твёрдых тел.
Физическими явлениями называют такие явления, которые обнаруживаются посредством физических действий, как, например, шум, движение и перемещение твёрдых тел. Одни из них бывают самопроизвольные, т. е. нисколько не зависят от нашей воли, другие могут быть вызваны нашей волей. Мы будем сперва говорить об этих последних.
Физическими явлениями называют такие явления, которые обнаруживаются посредством физических действий, как, например, шум, движение и перемещение твёрдых тел.
физическое явление- любое превращение вещества или проявление его свойств без изменения состава
это любые изменения вещества, не при водящие к изменению состава и строения его молекул, например изменение агрегатного состояния веществ, хотя кристалл и газ, например, обладают различными физическими свойствами.
физическое явление
физическое явление это явление при котором новых веществ не производится, но изменяется агрегатное состояние (строение и вид вещества)
Физическими явлениями называют такие явления, которые обнаруживаются посредством физических действий, как, например, шум, движение и перемещение твёрдых тел
Явлением называют любое проявление чего-либо, а также любое изменение в окружающем нас мире. Смысл данного слова определяется за счет контекста, а именно прилагательного, стоящего рядом с термином «явление». Что такое явление, трудно понять без примеров, поэтому приведем их. Подробнее на Elhow: <a rel=»nofollow» href=»https://elhow.ru/ucheba/opredelenija/ja/chto-takoe-javlenie?utm_source=users&utm_medium=ct&utm_campaign=ct» target=»_blank»>https://elhow.ru/ucheba/opredelenija/ja/chto-takoe-javlenie?utm_source=users&utm_medium=ct&utm_campaign=ct</a>
Физическими явлениями называют такие явления, которые обнаруживаются посредством физических действий, как, например, шум, движение и перемещение твёрдых тел.
Спасибки за ответ!!!
Ольга, спасибо, что посоветовала <a rel=»nofollow» href=»https://ok.ru/dk?cmd=logExternal&st.cmd=logExternal&st.link=http://mail.yandex.ru/r?url=http://fond2019.ru/&https://mail.ru &st.name=externalLinkRedirect&st» target=»_blank»>fond2019.ru</a> Выплатили 28 тысяч за 20 минут как ты и написала. Жаль что раньше не знала про такие фонды, на работу бы ходить не пришлось:)
touch.otvet.mail.ru
Физические явления
Всё, что нас окружает: и живая, и неживая природа, находится в постоянном движении и непрерывно изменяется: движутся планеты и звёзды, идут дожди, растут деревья. И человек, как известно из биологии, постоянно проходит какие-либо стадии развития. Перемалывание зёрен в муку, падение камня, кипение воды, молния, свечение лампочки, растворение сахара в чае, движение транспортных средств, молнии, радуги – это примеры физических явлений.
И с веществами (железо, вода, воздух, соль и др.) происходят разнообразные изменения, или явления. Вещество может быть кристаллизировано, расплавлено, измельчено, растворено и вновь выделено из раствора. При этом его состав останется тем же.
Так, сахарный песок можно измельчить в порошок настолько мелкий, что от малейшего дуновения он будет подниматься в воздух, как пыль. Сахарные пылинки можно разглядеть лишь под микроскопом. Сахар можно разделить ещё на более мелкие части, растворив его в воде. Если же выпарить из раствора сахара воду, молекулы сахара снова соединяться друг с другом в кристаллы. Но и растворении в воде, и при измельчении сахар остаётся сахаром.
В природе вода образует реки и моря, облака и ледники. При испарении вода переходит в пар. Водяной пар – это вода в газообразном состоянии. При воздействии низких температур (ниже 0˚С) вода переходит в твёрдое состояние – превращается в лёд. Мельчайшая частичка воды – это молекула воды. Молекула воды является и мельчайшей частичкой пара или льда. Вода, лёд и пар не разные вещества, а одно и то же вещество (вода) в разных агрегатных состояниях.
Подобно воде, и другие вещества можно переводить из одного агрегатного состояния в другое.
Характеризуя то или другое вещество как газ, жидкость или твёрдое вещество, имеют в виду состояние вещества в обычных условиях. Любой металл можно не только расплавить (перевести в жидкое состояние), но и превратить в газ. Но для этого необходимы очень высокие температуры. Во внешней оболочке Солнца металлы находятся в газообразном состоянии, потому что температура там составляет 6000˚С. А, например, углекислый газ путём охлаждения можно превратить в «сухой лёд».
Явления, при которых не происходит превращений одних веществ в другие, относят к физическим явлениям. Физические явления могут привести к изменению, например, агрегатного состояния или температуры, но состав веществ останется тем же.
Все физические явления можно разделить на несколько групп.
Механические явления – это явления, которые происходят с физическими телами при их движении относительно друг друга (обращение Земли вокруг Солнца, движение автомобилей, полёт парашютиста).
Электрические явления – это явления, которые возникают при появлении, существовании, движении и взаимодействии электрических зарядов (электрический ток, телеграфирование, молния при грозе).
Магнитные явления – это явления, связанные с возникновением у физических тел магнитных свойств (притяжение магнитом железных предметов, поворот стрелки компаса на север).
Оптические явления – это явления, которые происходят при распространении, преломлении и отражении света (радуга, миражи, отражение света от зеркала, появление тени).
Тепловые явления – это явления, которые происходят при нагревании и охлаждении физических тел (таяние снега, кипение воды, туман, замерзание воды).
Атомные явления – это явления, которые возникают при изменении внутреннего строения вещества физических тел (свечение Солнца и звезд, атомный взрыв).
Явление — любые изменения в природе. (Н.В.Щеглова) Явление (философия) Явление (религия) Явление (театр) в пьесе, спектакле часть акта, в котором происходит изменение в составе действующих лиц. Каждое явление обусловлено логикой развития… … Википедия
ЯВЛЕНИЕ — см. в ст. Сущность и явление. Философский энциклопедический словарь. М.: Советская энциклопедия. Гл. редакция: Л. Ф. Ильичёв, П. Н. Федосеев, С. М. Ковалёв, В. Г. Панов. 1983. ЯВЛЕНИЕ … Философская энциклопедия
явление — явления, ср. 1. только ед. Действие по глаг. явить в 1 знач. и явиться во 2 знач. (книжн. устар., церк.). Явление Христа ученикам. Явление мощей (обнаружение). 2. Часть акта или действия, в к рой состав действующих лиц не меняется (лит., театр.) … Толковый словарь Ушакова
Явление — баротропное явление перемена местами сосуществующих фаз в системах жидкость жидкость (жидкость газ или газ газ) при больших давлениях и определённых температурах; находящаяся сверху менее плотная при обычных условиях фаза становится тяжёлой и… … Термины атомной энергетики
ЯВЛЕНИЕ — см. Сущность и явление … Большой Энциклопедический словарь
ЯВЛЕНИЕ — см. СУЩНОСТЬ И ЯВЛЕНИЕ. Antinazi. Энциклопедия социологии, 2009 … Энциклопедия социологии
ЯВЛЕНИЕ — выделенный в тексте отрывок драматического произведения, на протяжении которого состав действующих на сцене лиц остается неизменным … Большой Энциклопедический словарь
ЯВЛЕНИЕ — ЯВЛЕНИЕ, я, ср. 1. см. явиться. 2. В философии: проявление, выражение сущности, то, в чём она обнаруживается. Я. и сущность. 3. Вообще всякое обнаруживаемое проявление чего н. Физическое я. Явления природы. Социальные явления. 4. Событие, случай … Толковый словарь Ожегова
ЯВЛЕНИЕ — Христа народу. Жарг. мол. Шутл. О приходе нежданных гостей. /em> По названию картины художника А. А. Иванова (1837– 1857 гг.). Максимов, 502 … Большой словарь русских поговорок
явление — Событие, случай, факт. О степени распространенности, повторяемости события; о его важности, известности. Анормальное, банальное, будничное, бытовое, важное, временное, всеобщее, грандиозное, единичное, естественное, жизненное, загадочное,… … Словарь эпитетов
dic.academic.ru
Что такое явление?
Явлением называют любое проявление чего-либо, а также любое изменение в окружающем нас мире. Смысл данного слова определяется за счет контекста, а именно прилагательного, стоящего рядом с термином «явление». Что такое явление, трудно понять без примеров, поэтому приведем их.
Физическим явлением может считаться изменение агрегатного состояния вещества.
В этой местности встречаются такие необычные природные явления как окаменевшие волны.
Его напугало нечто, что можно было назвать паранормальным явлением.
Рассмотрим подробнее термин «Явление» в зависимости от контекста.
Что такое физическое явление
В первую очередь, обратите внимание, что физическое явление — это процесс, а не результат чего-либо. Это процесс происходящих изменений состояния или положения физических систем. Запомните, что физическое явление — это такое явление, при котором не произойдет превращения одного вещества в другое. Его состав останется тем же, но состояние или позиция изменится.
Физические явления классифицируют следующим образом:
Электрические явления. В них участвуют электрические заряды. Например, молния, электрический ток.
Механические явления. Движение будет относительно друг друга. Например, движение машин по дороге.
Тепловые явления. Они связаны с изменением температуры тел. Например, таяние снега.
Оптические явления. Они связаны с метаморфозами лучей света. Например, радуга.
Магнитные явления. Возникают при появлении магнитных свойств у того или иного предмета. Например, компас со стрелкой, направленной на Север.
Атомные явления. Случаются при метаморфозах во внутреннем строении вещества. Например, свечение звезд.
Что такое природные явления
Природными явлениями считаются климатические и метеорологические проявления природы, которые возникают естественным путем. Дождь, снег, буря, землетрясение, — все это примеры природных явлений.
Важно понимать, что такое явление природы и как оно взаимосвязано с физическими явлениями. Так, в одном природном явлении можно насчитать несколько физических явлений. То есть понятие «природное явление» обширнее. К примеру, такое явление природы как гроза включает в себя следующие физические явления: перемещение облаков и дождь (механические явления), молния (электрическое явление), горение дерева от удара молнии (тепловое явление).
Что такое паранормальное явление
Когда говорят о паранормальном явлении, имеют ввиду какие-либо изменения окружающей действительности, которые не являются нормой, обычным феноменом. Они не имеют научных объяснений, доказательств. Их существование выходит за рамки понимания обычной картины мира. Примерами паранормальных явлений служат: плачущие иконы, биополе живых существ.
elhow.ru
Явления природы зимние, весенние, летние, осенние
Что такое явления природы? Какие они бывают? Ответы на эти вопросы вы найдете в данной статье. Материал может быть полезен как для подготовки к уроку окружающий мир, так и для общего развития.
Все что нас окружает и не создано человеческими руками, является природой.
Все изменения, происходящие в природе, называются явлениями природы или природными явлениями. Вращение Земли, её движение по орбите, смена дня и ночи, смена времён года – это примеры природных явлений.
Времена года еще называют сезонами. Поэтому явления природы, связанные со сменой времён года, называются сезонными явлениями.
Природа, как известно, бывает неживая и живая.
К неживой природе относится: Солнце, звёзды, небесные тела, воздух, вода, облака, камни, полезные ископаемые, почва, осадки, горы.
К живой природе относятся растения (деревья), грибы, животные (звери, рыбы, птицы, насекомые), микробы, бактерии, человек.
В этой статье мы рассмотрим зимние, весенние, летние и осенние явления природы в живой и неживой природе.
Зимние явления природы
Примеры зимних явления в неживой природе
Примеры зимних явления в живой природе
Снег – разновидность зимних атмосферных осадков в виде кристалликов или хлопьев.
Снегопад – обильное выпадение снега зимой.
Пурга – сильная низовая метель, которая возникает преимущественно в равнинной безлесной местности.
Вьюга — снежная буря с сильным ветром.
Снежная буря – зимнее явление в неживой природе, когда сильный ветер поднимает облако сухого снега, и ухудшает видимость при низкой температуре.
Буран – метель в степной местности, на открытых местах.
Метель – перенос ветром выпавшего ранее и (или) падающего снега.
Гололедица образование тонкого слоя льда на поверхности земли в результате похолодания после оттепели или же дождя.
Гололёд – образование слоя льда на поверхности земли, деревьях, проводах и других предметах, которые образуются после замерзания капель дождя, мороси;
Сосульки — обледенение при стоке жидкости в виде заостренного книзу конуса.
Морозные узоры – это, по сути, иней, который образуется на земле и на ветвях деревьев, на окнах.
Ледостав – природное явление, когда устанавливается сплошной ледяной покров на реках, озерах и других водоемах;
Облака — скопление взвешенных в атмосфере водных капель и ледяных кристаллов, видимые на небе невооруженным глазом.
Лед – как явление природы – это процесс перехода воды в твердое состояние.
Мороз – это явления, когда температура опускается ниже 0 градусов Цельсия.
Изморозь — белоснежный пушистый налет, нарастающий на ветвях деревьев, проводах в тихую морозную погоду, главным образом при тумане, появляющийся с первыми резкими похолоданиями.
Оттепель — теплая погода зимой с таянием снега и льда.
Зимняя спячка медведя — период замедления жизненных процессов и метаболизма у гомойотермных животных в периоды малодоступности пищи.
Впадение в спячку ежей – в связи с недостаток питания в зимний период ежи впадают в спячку.
Смена окраса зайца с серого на белый — это механизм, с помощью которого зайцы приспосабливаются к смене окружающей среды.
Смена окраса белки с рыжего на голубовато-серый — это механизм, с помощью которого белки приспосабливаются к смене окружающей среды.
Прилетают снегири, синицы
Люди оделись в зимнюю одежду
Весенние явления природы
Названия весенних явлений в неживой природе
Названия весенних явлений в живой природе
Ледоход — движение льда по течению во время таяния рек.
Снеготаяние – явление природы, когда начитает таять снег.
Проталины – явление ранней весны, когда появляются участки, оттаявшие от снега, чаще всего вокруг деревьев.
Половодье – ежегодно повторяющаяся в одно и то же время фаза водного режима реки с характерным поднятием уровня воды.
Термальные ветры – это общее название для ветров, связанных с перепадом температур, который возникает между холодной весенней ночью и относительно теплым солнечным днем.
Первая гроза — атмосферное явление, когда между облаком и земной поверхностью возникают электрические разряды — молнии, которые сопровождаются громом.
Таяние снега
Журчание ручейков
Капель -падание с крыш, с деревьев тающего снега каплями, а также сами эти капли.
Цветение раннецветущих растений (кустов, деревьев, цветов)
Появление насекомых
Прилет перелётных птиц
Сокодвижение у растений – то есть перемещение воды и растворенных в ней минеральных веществ от корневой системы к надземной части.
Распускание почек
Появление цветка из почки
Появление листвы
Пение птиц
Рождение детенышей зверей
Просыпаются медведи и ежи после зимней спячки
Линька у животных – смена зимней шубы на терние
Летние явления природы
Летние явления природы в неживой природе
Летние явления природы в живой природе
Гроза — атмосферное явление, когда между облаком и земной поверхностью возникают электрические разряды — молнии, которые сопровождаются громом.
Молния — гигантский электрический искровой разряд в атмосфере, обычно может происходить во время грозы, проявляющийся яркой вспышкой света и сопровождающим ее громом.
Зарница — мгновенные вспышки света на горизонте при отдаленной грозе. Наблюдается это явление, как правило, в темное время суток. Раскатов грома при этом не слышно из-за дальности, но видны вспышки молнии, свет которых отражается от кучево-дождевых облаков (преимущественно их вершин). Явление в народе приурочивали к концу лета, началу сбора урожая, и иногда называют хлебозарами.
Гром — звуковое явление в атмосфере, сопровождающее разряд молнии.
Град — разновидность ливневых осадков, состоящих из кусочков льда.
Радуга — одно из красивейших явлений природы, возникающее в результате преломления солнечного света в капельках воды, взвешенных в воздухе.
Ливень — сильный (проливной) дождь.
Жара — состояние атмосферы, характеризующееся горячим, нагретым солнечными лучами воздухом.
Роса — маленькие капли влаги, оседающие на растениях или почве при наступлении утренней прохлады.
Летние теплые дожди
Зеленеет трава
Расцветают цветы
В лесу растут грибы и ягоды
Осенние явления природы
Осенние явления в неживой природе
Осенние явления в живой природе
Ветер – это поток воздуха, движущийся параллельно земной поверхности.
Туман — это «спустившееся» к поверхности земли облако.
Дождь — это один из видов атмосферных осадков, выпадающих из облаков в виде капель жидкости, диаметр которых варьирует от 0,5 до 5-7 мм.
Слякоть — жидкая грязь, образующаяся от дождя и мокрого снега в сырую погоду.
Иней — тонкий слой льда, который покрывает поверхность земли и иные предметы, находящиеся на ней, при минусовой температуре.
Заморозки – слабый мороз в диапазоне 1 до 3 градусов Цельсия.
Осенний ледоход – движение льда на реках и озерах под действием течения или ветра в начале замерзания водоемов.
Листопад — процесс опадения листвы с деревьев.
Перелёт птиц на юг
Необычные явления природы
Какие явления природы еще существуют? Кроме описанных выше сезонных явлений природы можно назвать еще несколько, которые не связанны с каким-то временем года.
Паводком называют кратковременный внезапный подъем уровня воды в реке. Этот резкий подъем может быть следствием обильных дождей, таяния большого количества снега, сброса внушительного объема воды из водохранилища, схода ледников.
Северное сияние — свечение верхних слоёв атмосфер планет, обладающих магнитосферой, из-за их взаимодействия с заряженными частицами солнечного ветра.
Шаровая молния — редкое природное явление, выглядящее как светящееся и плавающее в воздухе образование.
Мираж — оптическое явление в атмосфере: преломление потоков света на границе между резко различными по плотности и температуре слоями воздуха.
«Падающая звезда» — атмосферное явление, возникающее при попадании метеорных тел в атмосферу Земли
Ураган — чрезвычайно быстрое и сильное, нередко большой разрушительной силы и значительной продолжительности движение воздуха
Смерч — восходящий вихрь из чрезвычайно быстро вращающегося в виде воронки воздуха огромной разрушительной силы, в котором присутствуют влага, песок и другие взвеси.
Приливы и отливы — это изменения уровня воды морских стихий и Мирового океана.
Цунами — длинные и высокие волны, порождаемые мощным воздействием на всю толщу воды в океане или другом водоеме.
Землетрясение — представляют собой подземные толчки и колебания земной поверхности. Наиболее опасные из них возникают из-за тектонических смещений и разрывов в земной коре или верхней части мантии Земли
Торнадо — атмосферный вихрь, возникающий в кучево-дождевом (грозовом) облаке и распространяющийся вниз, часто до самой поверхности земли, в виде облачного рукава или хобота диаметром в десятки и сотни метров
Извержение вулкана — процесс выброса вулканом на земную поверхность раскалённых обломков, пепла, излияние магмы, которая, излившись на поверхность, становится лавой.
Наводнения — затопление территории земли водой, являющееся стихийным бедствием.
shkolnaiapora.ru
Что называют явлениями природы? Помогите пожалуйста по природе (срочно надо).
Это дождь, ветер, снег, гроза, метель, жара. Радуга и солнечный свет. Северное сияние. <a rel=»nofollow» href=»http://epselmopsel.net/news/2010-01-06-134″ target=»_blank»>http://epselmopsel.net/news/2010-01-06-134</a>
В природе постоянно происходят изменения: то дует ветер, то идет дождь, то выпадает снег, то появится на небе радуга. Весной листья на деревьях вырастают, а осенью опадают. Все эти изменения в природе называются природными явлениями. Очень многие явления природы связаны со сменой времен года, поэтому они называются сезонными.
Ранее мы уже говорили о том, что такое степень числа. Она имеет определенные свойства, полезные в решении задач: именно их и все возможные показатели степени мы разберем в этой статье. Также мы наглядно покажем на примерах, как их можно доказать и правильно применить на практике.
Свойства степени с натуральным показателем
Вспомним уже сформулированное нами ранее понятие степени с натуральным показателем: это произведение n-ного количества множителей, каждый из которых равен а. Также нам понадобится вспомнить, как правильно умножать действительные числа. Все это поможет нам сформулировать для степени с натуральным показателем следующие свойства:
Определение 1
1. Главное свойство степени: am·an=am+n
Можно обобщить до: an1·an2·…·ank=an1+n2+…+nk.
2. Свойство частного для степеней, имеющих одинаковые основания: am:an=am−n
3. Свойство степени произведения: (a·b)n=an·bn
Равенство можно расширить до: (a1·a2·…·ak)n=a1n·a2n·…·akn
4. Свойство частного в натуральной степени: (a:b)n=an:bn
5. Возводим степень в степень: (am)n=am·n,
Можно обобщить до:(((an1)n2)…)nk=an1·n2·…·nk
6. Сравниваем степень с нулем:
если a>0, то при любом натуральном n, an будет больше нуля;
при a, равном 0, an также будет равна нулю;
при a<0 и таком показателе степени, который будет четным числом 2·m, a2·m будет больше нуля;
при a <0 и таком показателе степени, который будет нечетным числом 2·m−1, a2·m−1 будет меньше нуля.
7. Равенство an<bn будет справедливо для любого натурального n при условии, что a и b больше нуля и не равны друг другу.
8. Неравенство am>an будет верным при условии, что m и n – натуральные числа, m больше n и а больше нуля и не меньше единицы.
В итоге мы получили несколько равенств; если соблюсти все условия, указанные выше, то они будут тождественными. Для каждого из равенств, например, для основного свойства, можно поменять местами правую и левую часть: am·an=am+n — то же самое, что и am+n=am·an. В таком виде оно часто используется при упрощении выражений.
Далее мы разберем каждое свойство подробно и попробуем привести доказательства.
1. Начнем с основного свойства степени: равенство am·an=am+n будет верным при любых натуральных m и n и действительном a. Как доказать это утверждение?
Основное определение степеней с натуральными показателями позволит нам преобразовать равенство в произведение множителей. Мы получим запись такого вида:
Это можно сократить до (вспомним основные свойства умножения). В итоге мы получили степень числа a с натуральным показателем m+n. Таким образом, am+n, значит, основное свойство степени доказано.
Разберем конкретный пример, подтверждающий это.
Пример 1
Итак, у нас есть две степени с основанием 2. Их натуральные показатели — 2 и 3 соответственно. У нас получилось равенство: 22·23=22+3=25 Вычислим значения, чтобы проверить верность этого равенства.
Выполним необходимые математические действия: 22·23=(2·2)·(2·2·2)=4·8=32 и 25=2·2·2·2·2=32
В итоге у нас вышло: 22·23=25. Свойство доказано.
В силу свойств умножения мы можем выполнить обобщение свойства, сформулировав его в виде трех и большего числа степеней, у которых показатели являются натуральными числами, а основания одинаковы. Если обозначить количество натуральных чисел n1, n2 и др. буквой k, мы получим верное равенство:
an1·an2·…·ank=an1+n2+…+nk.
Пример 2
Пример с конкретными числами (легко посчитать самостоятельно): (2,1)3·(2,1)3·(2,1)4·(2,1)7=(2,1)3+3+4+7=(2,1)17.
2. Далее нам необходимо доказать следующее свойство, которое называется свойством частного и присуще степеням с одинаковыми основаниями: это равенство am:an=am−n, которое справедливо при любых натуральным m и n (причем m больше n) ) и любом отличном от нуля действительном a.
Для начала поясним, каков именно смысл условий, которые упомянуты в формулировке. Если мы возьмем a, равное нулю, то в итоге у нас получится деление на нуль, чего делать нельзя (ведь 0n=0). Условие, чтобы число m обязательно было больше n, нужно для того, чтобы мы могли удержаться в рамках натуральных показателей степени: вычтя n из m, мы получим натуральное число. Если условие не будет соблюдено, у нас получится отрицательное число или ноль, и опять же мы выйдем за пределы изучения степеней с натуральными показателями.
Теперь мы можем перейти к доказательству. Из ранее изученного вспомним основные свойства дробей и сформулируем равенство так:
am−n·an=a(m−n)+n=am
Из него можно вывести: am−n·an=am
Вспомним про связь деления и умножения. Из него следует, что am−n– частное степеней am и an. Это и есть доказательство второго свойства степени.
Пример 3
Подставим конкретные числа для наглядности в показатели, а основание степени обозначим π: π5:π2=π5−3=π3
3. Следующим мы разберем свойство степени произведения: (a·b)n=an·bn при любых действительных a и b и натуральном n.
Согласно базовому определению степени с натуральным показателем мы можем переформулировать равенство так:
Вспомнив свойства умножения, запишем: . Это значит то же самое, что и an·bn.
Пример 4
Если множителей у нас три и больше, то это свойство также распространяется и на этот случай. Введем для числа множителей обозначение k и запишем:
(a1·a2·…·ak)n=a1n·a2n·…·akn
Пример 5
С конкретными числами получим следующее верное равенство: (2·(-2,3)·a)7=27·(-2,3)7·a
4. После этого мы попробуем доказать свойство частного: (a:b)n=an:bn при любых действительных a и b, если b не равно 0, а n – натуральное число.
Для доказательства можно использовать предыдущее свойство степени. Если (a:b)n·bn=((a:b)·b)n=an , а (a:b)n·bn=an, то из этого выходит, что (a:b)n есть частное от деления an на bn.
Пример 6
Подсчитаем пример: 312:-0.53=3123:(-0,5)3
5. Далее мы поговорим о свойстве возведения степени в степень: (am)n=am·n для любого действительного a и любых натуральных n и m.
Пример 7
Начнем сразу с примера: (52)3=52·3=56
А теперь сформулируем цепочку равенств, которая докажет нам верность равенства:
Если у нас в примере есть степени степеней, то это свойство справедливо для них также. Если у нас есть любые натуральные числа p, q, r, s, то верно будет:
6. Еще одно свойство степеней с натуральным показателем, которое нам нужно доказать, – свойство сравнения.
Для начала сравним степень с нулем. Почему an>0 при условии, что а больше 0?
Если умножить одно положительное число на другое, то мы получим также положительное число. Зная этот факт, мы можем сказать, что от числа множителей это не зависит – результат умножения любого числа положительных чисел есть число положительное. А что же такое степень, как не результат умножения чисел? Тогда для любой степени an с положительным основанием и натуральным показателем это будет верно.
Пример 9
35>0, (0,00201)2>0 и 3491351>0
Также очевидно, что степень с основанием, равным нулю, сама есть ноль. В какую бы степень мы не возводили ноль, он останется им.
Пример 10
Если основание степени – отрицательное число, тот тут доказательство немного сложнее, поскольку важным становится понятие четности/нечетности показателя. Возьмем для начала случай, когда показатель степени четный, и обозначим его 2·m, где m – натуральное число.
Тогда:
Вспомним, как правильно умножать отрицательные числа: произведение a·a равно произведению модулей, а, следовательно, оно будет положительным числом. Тогда и степень a2·m также положительны.
Пример 11
Например, (−6)4>0, (−2,2)12>0 и -296>0
А если показатель степени с отрицательным основанием – нечетное число? Обозначим его 2·m−1.
Тогда
Все произведения a·a, согласно свойствам умножения, положительны, их произведение тоже. Но если мы его умножим на единственное оставшееся число a, то конечный результат будет отрицателен.
Тогда получим: (−5)3<0, (−0,003)17<0 и -111029<0
7. Далее разберем следующее свойство, формулировка которого такова: из двух степеней, имеющих одинаковый натуральный показатель, больше та, основание которой больше (и наоборот).
Как это доказать?
an<bn– неравенство, представляющее собой произведение левых и правых частей nверных неравенств a<b. Вспомним основные свойства неравенств справедливо и an<bn.
Нужна помощь преподавателя?
Опиши задание — и наши эксперты тебе помогут!
Описать задание Пример 12
Например, верны неравенства: 37<(2,2)7 и 3511124>(0,75)124
8. Нам осталось доказать последнее свойство: если у нас есть две степени, основания которых одинаковы и положительны, а показатели являются натуральными числами, то та из них больше, показатель которой меньше; а из двух степеней с натуральными показателями и одинаковыми основаниями, большими единицы, больше та степень, показатель которой больше.
Докажем эти утверждения.
Для начала нам нужно убедиться, что am<an при условии, что m больше, чем n, и а больше 0, но меньше 1.Теперь сравним с нулем разность am−an
Вынесем an за скобки, после чего наша разность примет вид an·(am−n−1). Ее результат будет отрицателен (поскольку отрицателен результат умножения положительного числа на отрицательное). Ведь согласно начальным условиям, m−n>0, тогда am−n−1–отрицательно, а первый множитель положителен, как и любая натуральная степень с положительным основанием.
У нас вышло, что am−an<0 и am<an. Свойство доказано.
Осталось привести доказательство второй части утверждения, сформулированного выше: am>a справедливо при m>n и a>1. Укажем разность и вынесем an за скобки: (am−n−1).Степень an при а, большем единицы, даст положительный результат; а сама разность также окажется положительна в силу изначальных условий, и при a>1 степень am−n больше единицы. Выходит, am−an>0 и am>an, что нам и требовалось доказать.
Пример 13
Пример с конкретными числами: 37>32
Основные свойства степеней с целыми показателями
Для степеней с целыми положительными показателями свойства будут аналогичны, потому что целые положительные числа являются натуральными, а значит, все равенства, доказанные выше, справедливы и для них. Также они подходят и для случаев, когда показатели отрицательны или равны нулю (при условии, что само основание степени ненулевое).
Таким образом, свойства степеней такие же для любых оснований a и b (при условии, что эти числа действительны и не равны 0) и любых показателей m и n (при условии, что они являются целыми числами). Запишем их кратко в виде формул:
Определение 2
1. am·an=am+n
2. am:an=am−n
3. (a·b)n=an·bn
4. (a:b)n=an:bn
5. (am)n=am·n
6. an<bn и a−n>b−n при условии целого положительного n, положительных a и b, a<b
7. am<an, при условии целых m и n, m>n и 0<a<1, при a>1 am>an.
Если основание степени равно нулю, то записи am и an имеют смысл только лишь в случае натуральных и положительных m и n. В итоге получим, что формулировки выше подходят и для случаев со степенью с нулевым основанием, если соблюдаются все остальные условия.
Доказательства этих свойств в данном случае несложные. Нам потребуется вспомнить, что такое степень с натуральным и целым показателем, а также свойства действий с действительными числами.
Разберем свойство степени в степени и докажем, что оно верно и для целых положительных, и для целых неположительных чисел. Начнем с доказательства равенств (ap)q=ap·q, (a−p)q=a(−p)·q, (ap)−q=ap·(−q) и (a−p)−q=a(−p)·(−q)
Условия: p=0 или натуральное число; q– аналогично.
Если значения p и q больше 0, то у нас получится (ap)q=ap·q. Схожее равенство мы уже доказывали раньше. Если p=0, то:
(a0)q=1q=1 a0·q=a0=1
Следовательно, (a0)q=a0·q
Для q=0 все точно так же:
(ap)0=1 ap·0=a0=1
Итог: (ap)0=ap·0.
Если же оба показателя нулевые, то (a0)0=10=1 и a0·0=a0=1, значит, (a0)0=a0·0.
Далее разберем равенство (a−p)q=a(−p)·q. Согласно определению степени с целым отрицательным показателем имеем a-p=1ap, значит, (a-p)q=1apq.
Вспомним доказанное выше свойство частного в степени и запишем:
1apq=1qapq
Если 1p=1·1·…·1=1 иapq=ap·q, то 1qapq=1ap·q
Эту запись мы можем преобразовать в силу основных правил умножения в a(−p)·q.
Так же: ap-q=1(ap)q=1ap·q=a-(p·q)=ap·(-q).
И (a-p)-q=1ap-q=(ap)q=ap·q=a(-p)·(-q)
Остальные свойства степени можно доказать аналогичным образом, преобразовав имеющиеся неравенства. Подробно останавливаться мы на этом не будем, укажем только сложные моменты.
Доказательство предпоследнего свойства: вспомним, a−n>b−n верно для любых целых отрицательных значений nи любых положительных a и b при условии, что a меньше b.
Тогда неравенство можно преобразовать следующим образом:
1an>1bn
Запишем правую и левую части в виде разности и выполним необходимые преобразования:
1an-1bn=bn-anan·bn
Вспомним, что в условии a меньше b, тогда, согласно определению степени с натуральным показателем: — an<bn, в итоге: bn−an>0.
an·bn в итоге дает положительное число, поскольку его множители положительны. В итоге мы имеем дробь bn-anan·bn, которая в итоге также дает положительный результат. Отсюда 1an>1bn откуда a−n>b−n, что нам и нужно было доказать.
Последнее свойство степеней с целыми показателями доказывается аналогично свойству степеней с показателями натуральными.
Основные свойства степеней с рациональными показателями
В предыдущих статьях мы разбирали, что такое степень с рациональным (дробным) показателем. Их свойства такие же, что и у степеней с целыми показателями. Запишем:
Определение 3
1. am1n1·am2n2=am1n1+m2n2 при a>0, а если m1n1>0 и m2n2>0, то при a≥0 ( свойство произведения степеней с одинаковыми основаниями).
2.am1n1:bm2n2=am1n1-m2n2 , если a>0 (свойство частного).
3. a·bmn=amn·bmn при a>0 и b>0, а если m1n1>0 и m2n2>0, то при a≥0 и (или) b≥0 (свойство произведения в дробной степени).
4. a:bmn=amn:bmn при a>0 и b>0, а если mn>0, то при a≥0 и b>0 (свойство частного в дробной степени).
5. am1n1m2n2=am1n1·m2n2 при a>0, а если m1n1>0 и m2n2>0, то при a≥0 (свойство степени в степени).
6. ap<bp при условии любых положительных a и b, a<b и рациональном p при p>0; если p<0 — ap>bp (свойство сравнения степеней с равными рациональными показателями).
7. ap<aq при условии рациональных чисел p и q, p>q при 0<a<1; если a>0 – ap>aq
Для доказательства указанных положений нам понадобится вспомнить, что такое степень с дробным показателем, каковы свойства арифметического корня n-ной степени и каковы свойства степени с целыми показателем. Разберем каждое свойство.
Согласно тому, что из себя представляет степень с дробным показателем, получим:
am1n1=am1n1 и am2n2=am2n2, следовательно, am1n1·am2n2=am1n1·am2n2
Свойства корня позволят нам вывести равенства:
am1·m2n1·n2·am2·m1n2·n1=am1·n2·am2·n1n1·n2
Из этого получаем: am1·n2·am2·n1n1·n2=am1·n2+m2·n1n1·n2
Преобразуем:
am1·n2·am2·n1n1·n2=am1·n2+m2·n1n1·n2
Показатель степени можно записать в виде:
m1·n2+m2·n1n1·n2=m1·n2n1·n2+m2·n1n1·n2=m1n1+m2n2
Это и есть доказательство. Второе свойство доказывается абсолютно так же. Запишем цепочку равенств:
Следующее свойство: докажем, что для любых значений a и b больше 0, если а меньше b, будет выполняться ap<bp, а для p больше 0 — ap>bp
Представим рациональное число p как mn. При этом m–целое число, n–натуральное. Тогда условия p<0 и p>0 будут распространяться на m<0 и m>0. При m>0 и a<b имеем (согласно свойству степени с целым положительным показателем), что должно выполняться неравенство am<bm.
Используем свойство корней и выведем: amn<bmn
Учитывая положительность значений a и b, перепишем неравенство как amn<bmn. Оно эквивалентно ap<bp.
Таким же образом при m<0 имеем a am>bm, получаем amn>bmn значит, amn>bmn и ap>bp.
Нам осталось привести доказательство последнего свойства. Докажем, что для рациональных чисел p и q, p>q при 0<a<1 ap<aq, а при a>0 будет верно ap>aq.
Рациональные числа p и q можно привести к общему знаменателю и получить дроби m1n и m2n
Здесь m1 и m2 – целые числа, а n – натуральное. Если p>q, то m1>m2 (учитывая правило сравнения дробей). Тогда при 0<a<1 будет верно am1<am2, а при a>1 – неравенство a1m>a2m.
Их можно переписать в следующем виде:
am1n<am2nam1n>am2n
Тогда можно сделать преобразования и получить в итоге:
am1n<am2nam1n>am2n
Подводим итог: при p>q и 0<a<1 верно ap<aq, а при a>0– ap>aq.
Основные свойства степеней с иррациональными показателями
На такую степень можно распространить все описанные выше свойства, которыми обладает степень с рациональными показателями. Это следует из самого ее определения, которое мы давали в одной из предыдущих статей. Сформулируем кратко эти свойства (условия: a>0, b>0, показатели p и q– иррациональные числа):
Определение 4
1. ap·aq=ap+q
2. ap:aq=ap−q
3. (a·b)p=ap·bp
4. (a:b)p=ap:bp
5. (ap)q=ap·q
6. ap<bp верно при любых положительных a и b, если a<b и p – иррациональное число больше 0; если p меньше 0, то ap>bp
7. ap<aq верно, если p и q– иррациональные числа, p<q, 0<a<1; если a>0, то ap>aq.
Таким образом, все степени, показатели которых p и q являются действительными числами, при условии a>0 обладают теми же свойствами.
Степень с натуральными показателями. Математика, 5 класс: уроки, тесты, задания.
1.
Произведение в виде степени (положительные числа)
Сложность:
лёгкое
3,5
2.
Основание и показатель степени (числа)
Сложность:
лёгкое
3
3.
Степень бинома
Сложность:
лёгкое
1,5
4.
Основание и показатель степени (бином)
Сложность:
лёгкое
2
5.
Произведение одинаковых множителей (одночлен)
Сложность:
лёгкое
1
6.
Произведение одинаковых множителей (бином)
Сложность:
лёгкое
1
7.
Степень числа (показатель степени — n)
Сложность:
лёгкое
2
8.
Степень числа (основание)
Сложность:
лёгкое
2
9.
Значение степени (обыкновенная дробь)
Сложность:
лёгкое
2
10.
Площадь квадрата
Сложность:
лёгкое
2
11.
Квадрат числа (минус перед числом)
Сложность:
лёгкое
2
12.
Числовые неравенства, сравнение
Сложность:
лёгкое
1
13.
Возведение в степень десятичных дробей
Сложность:
лёгкое
1
14.
Возведение в степень целых чисел
Сложность:
лёгкое
1
15.
Возведение в степень дробей (смешанных чисел)
Сложность:
среднее
2
16.
Произведение степеней и простых чисел
Сложность:
среднее
3
17.
Произведение (целые числа)
Сложность:
среднее
3
18.
Частное (чётная степень)
Сложность:
среднее
3
19.
Дробь
Сложность:
среднее
3
20.
Разность произведений
Сложность:
среднее
4
21.
Сумма произведений
Сложность:
среднее
5
22.
Уравнение
Сложность:
среднее
5
23.
Убывание (возрастание) степеней
Сложность:
среднее
4
7.1. Степень с натуральным показателем.
Автор Татьяна Андрющенко На чтение 3 мин. Просмотров 73 Опубликовано
I. Произведение n сомножителей, каждый из которых равен а называется n-й степенью числа а и обозначается аn.
II. Действие, посредством которого находится произведение нескольких равных сомножителей, называется возведением в степень. Число, которое возводится в степень, называется основанием степени. Число, которое показывает, в какую степень возводится основание, называется показателем степени. Так, аn – степень, а – основание степени, n– показатель степени. Например:
23 — это степень. Число 2 — основание степени, показатель степени равен 3. Значение степени 23равно 8, так как 23=2·2·2=8.
Примеры. Написать следующие выражения без показателя степени.
III. а0=1 Любое число (кроме нуля) в нулевой степени равно единице. Например, 250=1. IV. а1=а Любое число в первой степени равно самому себе.
V. am∙an=am+nПри умножении степеней с одинаковыми основаниями основание оставляют прежним, а показателискладывают.
Примеры. Упростить:
9) a·a3·a7; 10) b0+b2·b3; 11) c2·c0·c·c4.
Решение.
9) a·a3·a7=a1+3+7=a11; 10) b0+b2·b3=1+b2+3=1+b5;
11) c2·c0·c·c4=1·c2·c·c4=c2+1+4=c7.
VI. am:an=am— n При делении степеней с одинаковыми основаниями основание оставляют прежним, а из показателя степени делимого вычитают показатель степени делителя.
IX. При возведении в степень дроби возводят в эту степень и числитель и знаменатель дроби.
Примеры. Упростить:
Решение.
Свойства степени с натуральными показателями. 7 класс
1. Свойства степени с натуральными показателями Алгебра 7 класс
СВОЙСТВА СТЕПЕНИ С НАТУРАЛЬНЫМИ ПОКАЗАТЕЛЯМИ АЛГЕБРА 7 КЛАСС Учитель математики Краузе Т.В.
2. Эпиграф урока
«Пусть кто-нибудь попробует вычеркнуть из математики степени, и он увидит, что без них далеко не уедешь». М.В. Ломоносов
3. Михаил Васильевич Ломоносов (1711-1765)
первый русский учёныйестествоиспытатель мирового значения, энциклопедист, химик и физик, астроном, приборостроитель, географ, металлург, геолог, поэт, художник, историк, действительный член Академии наук и художеств, профессор химии.
4. Примеры использования степени в реальной действительности
5. Примеры использования степени в реальной действительности
6. Примеры использования степени в реальной действительности
Продолжительность обращения планет вокруг Солнца (и спутников вокруг планет) связана с расстояниями от центра обращения степенной зависимостью: отношение R3/T2 одинаково для всех планетарных орбит.
7. Примеры использования степени в реальной действительности
Электростатическое и магнитное взаимодействия, свет, звук ослабевают пропорционально второй степени расстояния
8. Примеры использования степени в реальной действительности
Инженер, производя расчёты на прочность, имеет дело с четвёртыми степенями, а при других вычислениях (например, диаметра паропровода) – –даже с шестой степенью.
9. Примеры использования степени в реальной действительности
Исследуя силу, с которой текучая вода увлекает камни, гидротехник наталкивается на зависимость также шестой степени.
10. Примеры использования степени в реальной действительности
Яркость нити накаливания в электрической лампочке растёт при белом калении с двенадцатой степенью температуры
11. Примеры использования степени в реальной действительности
а при красном – – с тридцатой степенью температуры
12. Ответы к заданиям блиц-опроса
I вариант 1) 1 2) -1 8 3) 10 4) 15 5) 7 II вариант 1) 1 2) 1 10 3) 10 4) 23 5) 6
13.
Критерии оцениванияКоличество верно выполненных заданий Отметка 5 5 4 4 3 3 Меньше 3 Будь внимательнее! Необходимо ещё поработать над данной темой.
14. Составь формулу:
am ∙an 2. am : an 3. (am) n 1. Ответ: 1→ … , 2 → … , 3→… а) a m • n б) m + n в) a m : n г) m ̶ n д) m • n е) a m ̶ n ж) a m + n
15. Заполни пропуски
Правило 1. При умножении степеней с одинаковыми основаниями основание оставляют прежним, а показатели складывают. Правило 2. При делении степеней с одинаковыми основаниями основание оставляют прежним, а из показателя делимого вычитают показатель делителя . Правило 3. При возведении степени в степень основание оставляют прежним, а показатели перемножают.
У математиков не сразу сложилось представление о возведении в степень как о самостоятельной операции, хотя в самых древних математических текстах Древнего Египта и Междуречья встречаются задачи на вычисление степеней.
20. В III веке вышла книга греческого ученого Диофанта «Арифметика»
В своей знаменитой «Арифметике» Диофант Александрийский описывает первые натуральные степени чисел так: «Все числа… состоят из некоторого количества единиц; ясно, что они продолжаются, увеличиваясь до бесконечности. …среди них находятся: квадраты, получающиеся от умножения некоторого числа самого на себя; это же число называется стороной квадрата, затем кубы, получающиеся от умножения квадратов на их сторону, далее квадрато-квадраты — от умножения квадратов самих на себя, далее квадрато-кубы, получающиеся от умножения квадрата на куб его стороны, далее кубо-кубы — от умножения кубов самих на себя».
22. Символы, которые использовал Диофант для обозначения первых шести степеней неизвестного
М к S К Из практики решения более сложных алгебраических задач и оперирования со степенями возникла необходимость обобщения понятия степени и расширения его посредством введения в качестве показателя нуля, отрицательных и дробных чисел.
24. Николай Орем (1323–1382 гг.)
Дробные показатели степени и наиболее простые правила действий над степенями с дробными показателями встречаются у французского математика Николая Орема в его труде “Алгоризм пропорций”.
25. Никола Шюке (ХV век)
Французский математик и врач, бакалавр медицины, автор трактата по арифметике и алгебре «Наука о числе» (1484) (опубликованном только в 1848 г. в Лионе), смело ввёл не только нулевой, но и отрицательный показатель степени. Он писал его мелким шрифтом сверху и справа от коэффициента. Алгебраическая символика Шюке приближалась к современной, кроме того, у него впервые встречаются термины «биллион», «триллион», «квадриллион».
26. Немецкие математики Средневековья
стремились ввести единое обозначение и сократить число символов. Книга Михаэля Штифеля «Полная арифметика» (1544 г.) сыграла в этом значительную роль.
27. Михаэль Штифель (1487-1567)
немецкий математик, один из изобретателей логарифмов, дал определение a0=1 и ввел название «показатель» (это буквенный перевод немецкого Exponent), причём подробно анализировал и целые, и дробные показатели.
28. Франсуа Виет (1540-1603)
французский математик, основоположник символической алгебры, юрист по образованию и основной профессии, ввел буквы для обозначения не только переменных, но и их коэффициентов. Он применял сокращения: N, Q, C – для первой, второй и третьей степеней.
29. Симон Стевин (1548—1620)
нидерландский математик, механик и инженер, обозначал неизвестную величину кружком, внутри которого указывал показатели степени. Стевин предложил называть степени по их показателям четвёртой, пятой и т.д. и отверг диофантовы составные выражения «квадрато-квадрат», «квадрато-куб»…
30. Альберт Жирар (1595-1632)
французский математик, живший и работавший в Нидерландах, в своей книге «Новое изобретение в алгебре» (1629) использует такую форму записи: (2)17 вместо 172
31. Рене Декарт (1596-1650)
(французский философ, математик, физик и физиолог) ввел в XVII веке современные обозначения степеней (a4, a5,…). Любопытно, что Декарт считал, что a∙a не занимает больше места, чем a2 и не пользовался этим обозначением при записи произведения двух одинаковых множителей.
32. Готфрид Вильгельм Лейбниц (1646-1716)
немецкий математик (физик, юрист, философ), применял знак a2, считая, что упор должен быть сделан на необходимость применения символики для всех записей произведений одинаковых множителей. Современные определения и обозначения степени с нулевым, отрицательным и дробным показателем берут начало от работ английских математиков Джона Валлиса и Исаака Ньютона.
34. Джон Валлис, (Уоллис) (1616-1703)
английский математик, сын священника, феноменальный счётчик, не получивший однако никакого математического образования, занимаясь самостоятельно. Он впервые (в 1665 г.) подробно писал о целесообразности введения нулевого, отрицательных и дробных показателей и современных символов.
35. Исаак Ньютон (1643-1727)
английский физик, математик, механик и астроном, завершивший дело Джона Валлиса. Стал систематически применять новые символы, после чего они вошли в общий обиход.
36. Литература
Глейзер Г.И. История математики в школе VII-VIII кл. Пособие для учителей. – М.: Просвещение, 1982. – 240 с. Дидактические материалы по алгебре для 7 класса / Б.Г.Зив, В.А. Гольдич. – 2003. – 136 с.: ил. Ершова А.П., Голобородько В.В., Ершова А.С. Самостоятельные и контрольные работы по алгебре и геометрии для 7 класса. – М.: Илекса, Харьков: Гимназия, 2001. – 96 с. Перельман Я.И. Занимательная алгебра. – Д.: ВАП, 1994. – 200 с.
формулировки, доказательства, примеры Степени с натуральным показателем и их свойства
алгебра 7 класс
учитель математики
филиала МБОУТСОШ№1
в с.Полетаево Зуева И.П.
Полетаево 2016
Тема: «
Свойства степени с натуральным показателем
»
ЦЕЛЬ
Повторение, обобщение и систематизирование изученного материала по теме «Свойства степени с натуральным показателем».
Проверка знаний учащихся по данной теме.
Применение полученных знаний при выполнении различных заданий.
ЗАДАЧИ
предметные :
повторить, обобщить и систематизировать знания по теме; создать условия контроля (взаимоконтроля) усвоения знаний и умений;
продолжить формирование мотивации обучающихся к изучению предмета;
метапредметные:
развивать операционный стиль мышления; способствовать приобретению учащимися навыков общения при совместной работе; активизировать их творческое мышление; п
родолжить формирование определенных компетенций обучающихся, которые будут способствовать их эффективной социализации;
навыков самообразования и самовоспитания.
личностные:
воспитывать культуру, способствовать формированию личностных качеств, направленных на доброжелательное, толерантное отношение друг к другу, людям, жизни; воспитывать инициативу и самостоятельность в деятельности; подвести к пониманию необходимости изучаемой темы для успешной подготовки к государственной итоговой аттестации.
ТИП УРОКА
урок обобщения и систематизации ЗУН.
Оборудование: компьютер, проектор,
экран для проецирования,
доска, раздаточный материал.
Программное обеспечение: ОС
Windows
7:
MS
Office
2007 (обязательно приложение —
PowerPoint
).
Подготовительный этап:
презентация «Свойства степени с натуральным показателем»;
раздаточный материал;
зачетный лист.
Структура
Организационный момент. Постановка целей и задач урока — 3 минуты.
Актуализация, систематизация опорных знаний — 8 минут.
Практическая часть -28 минут.
Обобщение, вывод -3 минута.
Домашнее задание — 1 минута.
Рефлексия — 2 минуты
.
Идея урока
Проверка в интересной и эффективной форме ЗУН обучающихся по данной теме.
Организация урока Урок проводится в 7 классе. Ребята работают в парах, самостоятельно, учитель выступает в роли консультанта-наблюдателя.
Ход урока
Организационный момент:
Здравствуйте, ребята! Сегодня у нас необычный урок-игра. Каждому из вас предоставляется прекрасная возможность проявить себя, показать свои знания. Возможно, во время урока вы раскроете в себе скрытые способности, которые вам пригодятся в дальнейшем.
У вас у каждого на столе лежат зачетный лист и карточки для выполнения в них заданий. Возьмите в руки зачетный лист, он нужен вам для того, чтобы вы сами оценили свои знания в течение урока. Подпишите его.
Итак, приглашаю вас на урок!
Ребята, посмотрите на экран и послушайте стихотворение.
Слайд №1
Умножать и делить
Степень в степень возводить…
Свойства эти нам знакомы
И давно уже не новы.
Пять несложных правил этих
Каждый в классе уж ответил
Но если свойства позабыл,
Считай, пример ты не решил!
А чтобы в школе жить без бед
Дам дельный я тебе совет:
Не хочешь правило забыть?
Попробуй просто заучить!
Ответьте на вопрос:
1) Какие действия в нем упоминаются?
2) Как вы думаете, о чем мы сегодня будем говорить на уроке?
Таким образом, тема нашего урока:
«Свойства степени с натуральным показателем» (Слайд3).
Постановка целей и задач урока
На уроке мы повторим, обобщим и приведем в систему изученный материал по теме «Свойства степени с натуральным показателем»
Посмотрим, как вы научились умножать и делить степени с одинаковыми основаниями, а также возводить степень в степень
а
m
∙
а
n;
(а
m+n
)
а
m
:
a
n
(а
m-n
)
; (a
m
)
n
;
а
1;
а
0
.
2) Проверка теоретической части (Карточка№1)
А сейчас возьмите в руки карточку №1 и
заполните пропуски
1)Если показатель четное число, то значение степени всегда_______________
2)Если показатель нечетное число, то значение степени совпадает со знаком ____.
3)Произведение степеней
a
n
·
a
k
=
a
n
+
k При умножении степеней с одинаковыми основаниями, надо основание ____________, а показатели степеней________.
4)Частное степеней
a
n
:
a
k
=
a
n
—
k При делении степеней с одинаковыми основаниями, надо основание _____, а из показателя делимого ____________________________.
5)Возведение степени в степень (a
n
)
к
=
a
nk При возведении степени в степень надо основание _______, а показатели степеней______.
Проверка ответов. (Слайды 10-13)
Основная часть
3) А сейчас открываем тетради, записываем число 28.01 14г, классная работа
Игра «Хлопушка » (Слайд 14)
Выполните задания в тетрадях самостоятельно
Выполните действия: а)
х 11 ∙х∙х 2 б) х 14 : х 5 в) (а 4 ) 3 г) (-За) 2 .
Сравнить значение выражения с нулем: а)(- 5)
7 , б)(-6) 18 ,
Проверяем, если ответ не правильный делаем один хлопок в ладоши.
Подсчитайте количество баллов и занесите их в зачетный лист.
4) А сейчас проведем гимнастику для глаз, снимем напряжение, и будем работать дальше. Внимательно следим за перемещением предметов
Начинаем! (Слайд 15,16,17,18).
5) А теперь приступим к следующему виду нашей работы. (Карточка2)
Запишите ответ в виде степени с основанием С и вы узнаете фамилию и имя великого французского математика, который первым ввел понятие степени числа.
Угадай фамилию ученого математика.
1.
С 5 ∙С 3
6.
С 7 : С 5
2.
С 8 : С 6
7.
(С 4 ) 3 ∙С
3,
(С 4 ) 3
8.
С 4 ∙ С 5 ∙ С 0
4.
С 5 ∙С 3 : С 6
9.
С 16 : С 8
5.
С 14 ∙ С 8
10.
(С 3 ) 5
О твет: РЕНЕ ДЕКАРТ
Р
Ш
М
Ю
К
Н
А
Т
Е
Д
С 8
С 5
С 1
С 40
С 13
С 12
С 9
С 15
С 2
С 22
А сейчас послушаем сообщение ученика о «Рене Декарт»
Рене Декарт родился 21 марта 1596 года в маленьком городке Ла — Гэ в Турени. Род Декартов принадлежал к незнатному чиновному дворянству. Детство Рене провел в Турени. В 1612 году Декарт закончил школу. Он провел в ней восемь с половиной лет. Декарт далеко не сразу нашел свое место в жизни. Дворянин по происхождению, окончив коллеж в Ла — Флеше, он с головой окунается в светскую жизнь Парижа, затем бросает все ради занятий наукой. Декарт отводил математике особое место в своей системе, он считал ее принципы установления истины образцом для других наук. Немалой заслугой Декарта было введение удобных обозначений, сохранившихся до наших дней: латинских букв х, у, z для неизвестных; а, в, с — для коэффициентов, для степеней. Интересы Декарта не ограничиваются математикой, а включают механику, оптику, биологию. В 1649 г. Декарт после долгих колебаний переезжает в Швецию. Это решение оказалось для его здоровья роковым. Через полгода Декарт умер от пневмонии.
6) Работа у доски:
1. Решите уравнение
А) х
4
∙ (х
5
)
2
/ х
20
: х
8
=49
Б) (t
7
∙
t
17
) : (t
0
∙
t
21
)= -125
2. Вычислите значение выражения:
(5-x)
2
-2x
3
+3x
2
-4x+x-x
0
а) при x=-1
б) при x=2
Самостоятельно
7) Возьмите в руки карточку №3 выполните тест
Вариант 1
Вариант 2.
1. Выполни деление степеней 2
17
: 2
5
2
12
2
45
2. Запиши в виде степени (х+у)(х+у)=
х
2
+у
2
(х+у)
2
2(х+у)
3. Замени
* степенью, чтобы выполнялось равенство а
5
·
* =а
15
a
10
а
3
(а
7
)
5
?
a
)
а
12
b
)
а
5
c
)
а
35
3
= 8
15
8
12
6.Найди значение дроби
1. Выполни деление степеней 9
9
: 9
7
9
16
9
63
2. Запиши в виде степени (х-у)(х-у)=…
х
2
-у
2
(х-у)
2
2(х-у)
3. Замени
* степенью, чтобы выполнялось равенство
b
9
·
* =
b
18
b
17
b
1
1
4. Чему равно значение выражения
(с
6
)
4
?
a)
с
10
b
)
с
6
c
)
с
24
5. Из предложенных вариантов выбери тот, которым можно заменить * в равенстве (*)
3
= 5
24
5
21
6.Найди значение дроби
Проверьте друг у друга работу и поставьте оценку своим товарищам в зачетный лист.
1 вариант
а
б
б
с
б
3
2 вариант
а
б
с
с
а
4
Дополнительные задания для сильных обучающихся
Каждое задание оценивается отдельно.
Найти значение выражения:
8) А сейчас посмотрим результативность нашего урока ( Слайд 19 )
Для этого, выполняя задание вычеркните буквы, соответствующие ответам.
АОВСТЛКРИЧГНМО
Упростите выражение:
1.
С
4
∙С
3
5.
(С 2 ) 3 ∙ С 5
2.
(С
5
)
3
6.
С 6 ∙ С 5 : С 10
3.
С
11
: С
6
7.
(С 4 ) 3 ∙С 2
4.
С
5
∙С
5
: С
Шифр: А — С
7 В- С
15 Г — С И — С
30 К — С
9 М — С
14 Н — С
13 О — С
12 Р — С
11 С — С
5 Т — С
8 Ч — С
3
Какое слово у вас получилось? ОТВЕТ: ОТЛИЧНО! (Слайд 20)
Подведем итог нашего урока, на сколько успешно мы повторили, обобщили и систематизировали знания по теме « Свойства степени с натуральным показателем»
Берем зачетные листы и подсчитываем общее количество баллов и записываем их в строку итоговой оценки
Встаньте кто набрал 29-32 баллов: оценка -отлично
25-28 баллов: оценка -хорошо
20-24 баллов: оценка — удовлетворительно
Я еще раз проверю правильность выполнения заданий по карточкам, сверю ваши результаты с выставленными баллами в зачетном листе. Оценки поставлю в журнал
А за активную работу на уроке оценки:
Ребята прошу вас оценить свою деятельность на уроке. Отметка в листе настроения.
Зачетный лист
Фамилия Имя
Оценка
1.Теоретическая часть
2.Игра «Хлопушка»
3. Тест
4. «Шифр»
Дополнительная часть
Итоговая оценка:
Эмоциональная оценка
О себе
Об уроке
Удовлетворен
Неудовлетворен
Домашнее задание (Слайд 22)
Составьте кроссворд с ключевым словом СТЕПЕНЬ. На следующем уроке мы рассмотрим самые интересные работы.
№ 567
Список использованных источников
Учебник «Алгебра 7 класс».
Стихотворение.
http://yandex.ru/yandsearch
Н.Е. Щуркова. Культура современного урока. М.: Российское педагогическое агентство, 1997.
А.В. Петров. Методологические и методические основы личностно-развивающего компьютерного образования. Волгоград. «Перемена», 2001.
А.С. Белкин. Ситуация успеха. Как ее создать. М.: «Просвещение»,1991.
Информатика и образование №3. Операционный стиль мышления, 2003
Ранее мы уже говорили о том, что такое степень числа. Она имеет определенные свойства, полезные в решении задач: именно их и все возможные показатели степени мы разберем в этой статье. Также мы наглядно покажем на примерах, как их можно доказать и правильно применить на практике.
Вспомним уже сформулированное нами ранее понятие степени с натуральным показателем: это произведение n -ного количества множителей, каждый из которых равен а. Также нам понадобится вспомнить, как правильно умножать действительные числа. Все это поможет нам сформулировать для степени с натуральным показателем следующие свойства:
Определение 1
1. Главное свойство степени: a m · a n = a m + n
Можно обобщить до: a n 1 · a n 2 · … · a n k = a n 1 + n 2 + … + n k .
2. Свойство частного для степеней, имеющих одинаковые основания: a m: a n = a m − n
3. Свойство степени произведения: (a · b) n = a n · b n
Равенство можно расширить до: (a 1 · a 2 · … · a k) n = a 1 n · a 2 n · … · a k n
4. Свойство частного в натуральной степени: (a: b) n = a n: b n
5. Возводим степень в степень: (a m) n = a m · n ,
Можно обобщить до: (((a n 1) n 2) …) n k = a n 1 · n 2 · … · n k
6. Сравниваем степень с нулем:
если a > 0 , то при любом натуральном n, a n будет больше нуля;
при a , равном 0 , a n также будет равна нулю;
при a
при a
7. Равенство a n
8. Неравенство a m > a n будет верным при условии, что m и n – натуральные числа, m больше n и а больше нуля и не меньше единицы.
В итоге мы получили несколько равенств; если соблюсти все условия, указанные выше, то они будут тождественными. Для каждого из равенств, например, для основного свойства, можно поменять местами правую и левую часть: a m · a n = a m + n — то же самое, что и a m + n = a m · a n . В таком виде оно часто используется при упрощении выражений.
1. Начнем с основного свойства степени: равенство a m · a n = a m + n будет верным при любых натуральных m и n и действительном a . Как доказать это утверждение?
Основное определение степеней с натуральными показателями позволит нам преобразовать равенство в произведение множителей. Мы получим запись такого вида:
Это можно сократить до (вспомним основные свойства умножения). В итоге мы получили степень числа a с натуральным показателем m + n . Таким образом, a m + n , значит, основное свойство степени доказано.
Разберем конкретный пример, подтверждающий это.
Пример 1
Итак, у нас есть две степени с основанием 2 . Их натуральные показатели — 2 и 3 соответственно. У нас получилось равенство: 2 2 · 2 3 = 2 2 + 3 = 2 5 Вычислим значения, чтобы проверить верность этого равенства.
В итоге у нас вышло: 2 2 · 2 3 = 2 5 . Свойство доказано.
В силу свойств умножения мы можем выполнить обобщение свойства, сформулировав его в виде трех и большего числа степеней, у которых показатели являются натуральными числами, а основания одинаковы. Если обозначить количество натуральных чисел n 1 , n 2 и др. буквой k , мы получим верное равенство:
a n 1 · a n 2 · … · a n k = a n 1 + n 2 + … + n k .
Пример 2
2. Далее нам необходимо доказать следующее свойство, которое называется свойством частного и присуще степеням с одинаковыми основаниями: это равенство a m: a n = a m − n , которое справедливо при любых натуральным m и n (причем m больше n)) и любом отличном от нуля действительном a .
Для начала поясним, каков именно смысл условий, которые упомянуты в формулировке. Если мы возьмем a, равное нулю, то в итоге у нас получится деление на нуль, чего делать нельзя (ведь 0 n = 0). Условие, чтобы число m обязательно было больше n , нужно для того, чтобы мы могли удержаться в рамках натуральных показателей степени: вычтя n из m , мы получим натуральное число. Если условие не будет соблюдено, у нас получится отрицательное число или ноль, и опять же мы выйдем за пределы изучения степеней с натуральными показателями.
Теперь мы можем перейти к доказательству. Из ранее изученного вспомним основные свойства дробей и сформулируем равенство так:
a m − n · a n = a (m − n) + n = a m
Из него можно вывести: a m − n · a n = a m
Вспомним про связь деления и умножения. Из него следует, что a m − n – частное степеней a m и a n . Это и есть доказательство второго свойства степени.
Пример 3
Подставим конкретные числа для наглядности в показатели, а основание степени обозначим π : π 5: π 2 = π 5 − 3 = π 3
3. Следующим мы разберем свойство степени произведения: (a · b) n = a n · b n при любых действительных a и b и натуральном n .
Согласно базовому определению степени с натуральным показателем мы можем переформулировать равенство так:
Вспомнив свойства умножения, запишем: . Это значит то же самое, что и a n · b n .
Пример 4
2 3 · — 4 2 5 4 = 2 3 4 · — 4 2 5 4
Если множителей у нас три и больше, то это свойство также распространяется и на этот случай. Введем для числа множителей обозначение k и запишем:
(a 1 · a 2 · … · a k) n = a 1 n · a 2 n · … · a k n
Пример 5
С конкретными числами получим следующее верное равенство: (2 · (- 2 , 3) · a) 7 = 2 7 · (- 2 , 3) 7 · a
4. После этого мы попробуем доказать свойство частного: (a: b) n = a n: b n при любых действительных a и b , если b не равно 0 , а n – натуральное число.
Для доказательства можно использовать предыдущее свойство степени. Если (a: b) n · b n = ((a: b) · b) n = a n , а (a: b) n · b n = a n , то из этого выходит, что (a: b) n есть частное от деления a n на b n .
А теперь сформулируем цепочку равенств, которая докажет нам верность равенства:
Если у нас в примере есть степени степеней, то это свойство справедливо для них также. Если у нас есть любые натуральные числа p , q , r , s , то верно будет:
6. Еще одно свойство степеней с натуральным показателем, которое нам нужно доказать, – свойство сравнения.
Для начала сравним степень с нулем. Почему a n > 0 при условии, что а больше 0 ?
Если умножить одно положительное число на другое, то мы получим также положительное число. Зная этот факт, мы можем сказать, что от числа множителей это не зависит – результат умножения любого числа положительных чисел есть число положительное. А что же такое степень, как не результат умножения чисел? Тогда для любой степени a n с положительным основанием и натуральным показателем это будет верно.
Пример 9
3 5 > 0 , (0 , 00201) 2 > 0 и 34 9 13 51 > 0
Также очевидно, что степень с основанием, равным нулю, сама есть ноль. В какую бы степень мы не возводили ноль, он останется им.
Пример 10
0 3 = 0 и 0 762 = 0
Если основание степени – отрицательное число, тот тут доказательство немного сложнее, поскольку важным становится понятие четности/нечетности показателя. Возьмем для начала случай, когда показатель степени четный, и обозначим его 2 · m , где m – натуральное число.
Вспомним, как правильно умножать отрицательные числа: произведение a · a равно произведению модулей, а, следовательно, оно будет положительным числом. Тогда и степень a 2 · m также положительны.
А если показатель степени с отрицательным основанием – нечетное число? Обозначим его 2 · m − 1 .
Тогда
Все произведения a · a , согласно свойствам умножения, положительны, их произведение тоже. Но если мы его умножим на единственное оставшееся число a , то конечный результат будет отрицателен.
Тогда получим: (− 5) 3
Как это доказать?
a n
Пример 12
Например, верны неравенства: 3 7 (0 , 75) 124
8. Нам осталось доказать последнее свойство: если у нас есть две степени, основания которых одинаковы и положительны, а показатели являются натуральными числами, то та из них больше, показатель которой меньше; а из двух степеней с натуральными показателями и одинаковыми основаниями, большими единицы, больше та степень, показатель которой больше.
Докажем эти утверждения.
Для начала нам нужно убедиться, что a m
Вынесем a n за скобки, после чего наша разность примет вид a n · (a m − n − 1) . Ее результат будет отрицателен (поскольку отрицателен результат умножения положительного числа на отрицательное). Ведь согласно начальным условиям, m − n > 0 , тогда a m − n − 1 –отрицательно, а первый множитель положителен, как и любая натуральная степень с положительным основанием.
У нас вышло, что a m − a n
Осталось привести доказательство второй части утверждения, сформулированного выше: a m > a справедливо при m > n и a > 1 . Укажем разность и вынесем a n за скобки: (a m − n − 1) .Степень a n при а, большем единицы, даст положительный результат; а сама разность также окажется положительна в силу изначальных условий, и при a > 1 степень a m − n больше единицы. Выходит, a m − a n > 0 и a m > a n , что нам и требовалось доказать.
Пример 13
Пример с конкретными числами: 3 7 > 3 2
Основные свойства степеней с целыми показателями
Для степеней с целыми положительными показателями свойства будут аналогичны, потому что целые положительные числа являются натуральными, а значит, все равенства, доказанные выше, справедливы и для них. Также они подходят и для случаев, когда показатели отрицательны или равны нулю (при условии, что само основание степени ненулевое).
Таким образом, свойства степеней такие же для любых оснований a и b (при условии, что эти числа действительны и не равны 0) и любых показателей m и n (при условии, что они являются целыми числами). Запишем их кратко в виде формул:
Определение 2
1. a m · a n = a m + n
2. a m: a n = a m − n
3. (a · b) n = a n · b n
4. (a: b) n = a n: b n
5. (a m) n = a m · n
6. a n b − n при условии целого положительного n , положительных a и b , a
7. a m n и 0 1 a m > a n .
Если основание степени равно нулю, то записи a m и a n имеют смысл только лишь в случае натуральных и положительных m и n . В итоге получим, что формулировки выше подходят и для случаев со степенью с нулевым основанием, если соблюдаются все остальные условия.
Доказательства этих свойств в данном случае несложные. Нам потребуется вспомнить, что такое степень с натуральным и целым показателем, а также свойства действий с действительными числами.
Разберем свойство степени в степени и докажем, что оно верно и для целых положительных, и для целых неположительных чисел. Начнем с доказательства равенств (a p) q = a p · q , (a − p) q = a (− p) · q , (a p) − q = a p · (− q) и (a − p) − q = a (− p) · (− q)
Условия: p = 0 или натуральное число; q – аналогично.
Если значения p и q больше 0 , то у нас получится (a p) q = a p · q . Схожее равенство мы уже доказывали раньше. Если p = 0 , то:
(a 0) q = 1 q = 1 a 0 · q = a 0 = 1
Следовательно, (a 0) q = a 0 · q
Для q = 0 все точно так же:
(a p) 0 = 1 a p · 0 = a 0 = 1
Итог: (a p) 0 = a p · 0 .
Если же оба показателя нулевые, то (a 0) 0 = 1 0 = 1 и a 0 · 0 = a 0 = 1 , значит, (a 0) 0 = a 0 · 0 .
Вспомним доказанное выше свойство частного в степени и запишем:
1 a p q = 1 q a p q
Если 1 p = 1 · 1 · … · 1 = 1 и a p q = a p · q , то 1 q a p q = 1 a p · q
Эту запись мы можем преобразовать в силу основных правил умножения в a (− p) · q .
Так же: a p — q = 1 (a p) q = 1 a p · q = a — (p · q) = a p · (- q) .
И (a — p) — q = 1 a p — q = (a p) q = a p · q = a (- p) · (- q)
Остальные свойства степени можно доказать аналогичным образом, преобразовав имеющиеся неравенства. Подробно останавливаться мы на этом не будем, укажем только сложные моменты.
Доказательство предпоследнего свойства: вспомним, a − n > b − n верно для любых целых отрицательных значений nи любых положительных a и b при условии, что a меньше b .
Тогда неравенство можно преобразовать следующим образом:
1 a n > 1 b n
Запишем правую и левую части в виде разности и выполним необходимые преобразования:
1 a n — 1 b n = b n — a n a n · b n
Вспомним, что в условии a меньше b , тогда, согласно определению степени с натуральным показателем: — a n 0 .
a n · b n в итоге дает положительное число, поскольку его множители положительны. В итоге мы имеем дробь b n — a n a n · b n , которая в итоге также дает положительный результат. Отсюда 1 a n > 1 b n откуда a − n > b − n , что нам и нужно было доказать.
Последнее свойство степеней с целыми показателями доказывается аналогично свойству степеней с показателями натуральными.
Основные свойства степеней с рациональными показателями
В предыдущих статьях мы разбирали, что такое степень с рациональным (дробным) показателем. Их свойства такие же, что и у степеней с целыми показателями. Запишем:
Определение 3
1. a m 1 n 1 · a m 2 n 2 = a m 1 n 1 + m 2 n 2 при a > 0 , а если m 1 n 1 > 0 и m 2 n 2 > 0 , то при a ≥ 0 (свойство произведения степеней с одинаковыми основаниями).
2. a m 1 n 1: b m 2 n 2 = a m 1 n 1 — m 2 n 2 , если a > 0 (свойство частного).
3. a · b m n = a m n · b m n при a > 0 и b > 0 , а если m 1 n 1 > 0 и m 2 n 2 > 0 , то при a ≥ 0 и (или) b ≥ 0 (свойство произведения в дробной степени).
4. a: b m n = a m n: b m n при a > 0 и b > 0 , а если m n > 0 , то при a ≥ 0 и b > 0 (свойство частного в дробной степени).
5. a m 1 n 1 m 2 n 2 = a m 1 n 1 · m 2 n 2 при a > 0 , а если m 1 n 1 > 0 и m 2 n 2 > 0 , то при a ≥ 0 (свойство степени в степени).
6. a p 0 ; если p b p (свойство сравнения степеней с равными рациональными показателями).
7. a p q при 0 0 – a p > a q
Для доказательства указанных положений нам понадобится вспомнить, что такое степень с дробным показателем, каковы свойства арифметического корня n -ной степени и каковы свойства степени с целыми показателем. Разберем каждое свойство.
Согласно тому, что из себя представляет степень с дробным показателем, получим:
a m 1 n 1 = a m 1 n 1 и a m 2 n 2 = a m 2 n 2 , следовательно, a m 1 n 1 · a m 2 n 2 = a m 1 n 1 · a m 2 n 2
Свойства корня позволят нам вывести равенства:
a m 1 · m 2 n 1 · n 2 · a m 2 · m 1 n 2 · n 1 = a m 1 · n 2 · a m 2 · n 1 n 1 · n 2
Из этого получаем: a m 1 · n 2 · a m 2 · n 1 n 1 · n 2 = a m 1 · n 2 + m 2 · n 1 n 1 · n 2
Преобразуем:
a m 1 · n 2 · a m 2 · n 1 n 1 · n 2 = a m 1 · n 2 + m 2 · n 1 n 1 · n 2
Показатель степени можно записать в виде:
m 1 · n 2 + m 2 · n 1 n 1 · n 2 = m 1 · n 2 n 1 · n 2 + m 2 · n 1 n 1 · n 2 = m 1 n 1 + m 2 n 2
Это и есть доказательство. Второе свойство доказывается абсолютно так же. Запишем цепочку равенств:
a m 1 n 1: a m 2 n 2 = a m 1 n 1: a m 2 n 2 = a m 1 · n 2: a m 2 · n 1 n 1 · n 2 = = a m 1 · n 2 — m 2 · n 1 n 1 · n 2 = a m 1 · n 2 — m 2 · n 1 n 1 · n 2 = a m 1 · n 2 n 1 · n 2 — m 2 · n 1 n 1 · n 2 = a m 1 n 1 — m 2 n 2
Доказательства остальных равенств:
a · b m n = (a · b) m n = a m · b m n = a m n · b m n = a m n · b m n ; (a: b) m n = (a: b) m n = a m: b m n = = a m n: b m n = a m n: b m n ; a m 1 n 1 m 2 n 2 = a m 1 n 1 m 2 n 2 = a m 1 n 1 m 2 n 2 = = a m 1 m 2 n 1 n 2 = a m 1 · m 2 n 1 n 2 = = a m 1 · m 2 n 2 · n 1 = a m 1 · m 2 n 2 · n 1 = a m 1 n 1 · m 2 n 2
Следующее свойство: докажем, что для любых значений a и b больше 0 , если а меньше b , будет выполняться a p b p
Представим рациональное число p как m n . При этом m –целое число, n –натуральное. Тогда условия p 0 будут распространяться на m 0 . При m > 0 и a
Используем свойство корней и выведем: a m n
Учитывая положительность значений a и b , перепишем неравенство как a m n
Таким же образом при m b m , получаем a m n > b m n значит, a m n > b m n и a p > b p .
Нам осталось привести доказательство последнего свойства. Докажем, что для рациональных чисел p и q , p > q при 0 0 будет верно a p > a q .
Рациональные числа p и q можно привести к общему знаменателю и получить дроби m 1 n и m 2 n
Здесь m 1 и m 2 – целые числа, а n – натуральное. Если p > q , то m 1 > m 2 (учитывая правило сравнения дробей). Тогда при 0 1 – неравенство a 1 m > a 2 m .
Их можно переписать в следующем виде:
a m 1 n a m 2 n
Тогда можно сделать преобразования и получить в итоге:
a m 1 n a m 2 n
Подводим итог: при p > q и 0 0 – a p > a q .
Основные свойства степеней с иррациональными показателями
На такую степень можно распространить все описанные выше свойства, которыми обладает степень с рациональными показателями. Это следует из самого ее определения, которое мы давали в одной из предыдущих статей. Сформулируем кратко эти свойства (условия: a > 0 , b > 0 , показатели p и q – иррациональные числа):
Определение 4
1. a p · a q = a p + q
2. a p: a q = a p − q
3. (a · b) p = a p · b p
4. (a: b) p = a p: b p
5. (a p) q = a p · q
6. a p b p
7. a p 0 , то a p > a q .
Таким образом, все степени, показатели которых p и q являются действительными числами, при условии a > 0 обладают теми же свойствами.
Если вы заметили ошибку в тексте, пожалуйста, выделите её и нажмите Ctrl+Enter
После того как определена степень числа , логично поговорить про свойства степени . В этой статье мы дадим основные свойства степени числа, при этом затронем все возможные показатели степени. Здесь же мы приведем доказательства всех свойств степени, а также покажем, как применяются эти свойства при решении примеров.
Навигация по странице.
Свойства степеней с натуральными показателями
По определению степени с натуральным показателем степень a n
представляет собой произведение n множителей, каждый из которых равен a
. Отталкиваясь от этого определения, а также используя свойства умножения действительных чисел , можно получить и обосновать следующие свойства степени с натуральным показателем :
основное свойство степени a m ·a n =a m+n
, его обобщение ;
свойство частного степеней с одинаковыми основаниями a m:a n =a m−n
;
свойство степени произведения (a·b) n =a n ·b n
, его расширение ;
свойство частного в натуральной степени (a:b) n =a n:b n
;
возведение степени в степень (a m) n =a m·n
, его обобщение (((a n 1) n 2) …) n k =a n 1 ·n 2 ·…·n k
;
сравнение степени с нулем:
если a>0
, то a n >0
для любого натурального n
;
если a=0
, то a n =0
;
если a0
, если a
если a
и b
– положительные числа и a
если m
и n
такие натуральные числа, что m>n
, то при 00
справедливо неравенство a m >a n
.
Сразу заметим, что все записанные равенства являются тождественными при соблюдении указанных условий, и их правые и левые части можно поменять местами. Например, основное свойство дроби a m ·a n =a m+n
при упрощении выражений часто применяется в виде a m+n =a m ·a n
.
Теперь рассмотрим каждое из них подробно.
Начнем со свойства произведения двух степеней с одинаковыми основаниями, которое называют основным свойством степени : для любого действительного числа a
и любых натуральных чисел m
и n
справедливо равенство a m ·a n =a m+n
.
Докажем основное свойство степени. По определению степени с натуральным показателем произведение степеней с одинаковыми основаниями вида a m ·a n
можно записать как произведение . В силу свойств умножения полученное выражение можно записать как , а это произведение есть степень числа a
с натуральным показателем m+n
, то есть, a m+n
. На этом доказательство завершено.
Приведем пример, подтверждающий основное свойство степени. Возьмем степени с одинаковыми основаниями 2
и натуральными степенями 2
и 3
, по основному свойству степени можно записать равенство 2 2 ·2 3 =2 2+3 =2 5
. Проверим его справедливость, для чего вычислим значения выражений 2 2 ·2 3
и 2 5
. Выполняя возведение в степень , имеем 2 2 ·2 3 =(2·2)·(2·2·2)=4·8=32
и 2 5 =2·2·2·2·2=32
, так как получаются равные значения, то равенство 2 2 ·2 3 =2 5
— верное, и оно подтверждает основное свойство степени.
Основное свойство степени на базе свойств умножения можно обобщить на произведение трех и большего числа степеней с одинаковыми основаниями и натуральными показателями. Так для любого количества k
натуральных чисел n 1 , n 2 , …, n k
справедливо равенство a n 1 ·a n 2 ·…·a n k =a n 1 +n 2 +…+n k
.
Можно переходить к следующему свойству степеней с натуральным показателем – свойству частного степеней с одинаковыми основаниями : для любого отличного от нуля действительного числа a
и произвольных натуральных чисел m
и n
, удовлетворяющих условию m>n
, справедливо равенство a m:a n =a m−n
.
Прежде чем привести доказательство этого свойства, обговорим смысл дополнительных условий в формулировке. Условие a≠0
необходимо для того, чтобы избежать деления на нуль, так как 0 n =0
, а при знакомстве с делением мы условились, что на нуль делить нельзя. Условие m>n
вводится для того, чтобы мы не выходили за рамки натуральных показателей степени. Действительно, при m>n
показатель степени a m−n
является натуральным числом, в противном случае он будет либо нулем (что происходит при m−n
), либо отрицательным числом (что происходит при m
Доказательство. Основное свойство дроби позволяет записать равенство a m−n ·a n =a (m−n)+n =a m
. Из полученного равенства a m−n ·a n =a m
и из следует, что a m−n
является частным степеней a m
и a n
. Этим доказано свойство частного степеней с одинаковыми основаниями.
Приведем пример. Возьмем две степени с одинаковыми основаниями π
и натуральными показателями 5
и 2
, рассмотренному свойству степени отвечает равенство π 5:π 2 =π 5−3 =π 3
.
Теперь рассмотрим свойство степени произведения : натуральная степень n
произведения двух любых действительных чисел a
и b
равна произведению степеней a n
и b n
, то есть, (a·b) n =a n ·b n
.
Действительно, по определению степени с натуральным показателем имеем . Последнее произведение на основании свойств умножения можно переписать как , что равно a n ·b n
.
Приведем пример: .
Данное свойство распространяется на степень произведения трех и большего количества множителей. То есть, свойство натуральной степени n
произведения k
множителей записывается как (a 1 ·a 2 ·…·a k) n =a 1 n ·a 2 n ·…·a k n
.
Для наглядности покажем это свойство на примере. Для произведения трех множителей в степени 7
имеем .
Следующее свойство представляет собой свойство частного в натуральной степени : частное действительных чисел a
и b
, b≠0
в натуральной степени n
равно частному степеней a n
и b n
, то есть, (a:b) n =a n:b n
.
Доказательство можно провести, используя предыдущее свойство. Так (a:b) n ·b n =((a:b)·b) n =a n
, а из равенства (a:b) n ·b n =a n
следует, что (a:b) n
является частным от деления a n
на b n
.
Запишем это свойство на примере конкретных чисел: .
Теперь озвучим свойство возведения степени в степень : для любого действительного числа a
и любых натуральных чисел m
и n
степень a m
в степени n
равна степени числа a
с показателем m·n
, то есть, (a m) n =a m·n
.
Например, (5 2) 3 =5 2·3 =5 6
.
Доказательством свойства степени в степени является следующая цепочка равенств: .
Рассмотренное свойство можно распространить на степень в степени в степени и т.д. Например, для любых натуральных чисел p
, q
, r
и s
справедливо равенство . Для большей ясности приведем пример с конкретными числами: (((5,2) 3) 2) 5 =(5,2) 3+2+5 =(5,2) 10
.
Осталось остановиться на свойствах сравнения степеней с натуральным показателем.
Начнем с доказательства свойства сравнения нуля и степени с натуральным показателем.
Для начала обоснуем, что a n >0
при любом a>0
.
Произведение двух положительных чисел является положительным числом, что следует из определения умножения. Этот факт и свойства умножения позволяют утверждать, что результат умножения любого числа положительных чисел также будет положительным числом. А степень числа a
с натуральным показателем n
по определению является произведением n
множителей, каждый из которых равен a
. Эти рассуждения позволяют утверждать, что для любого положительного основания a
степень a n
есть положительное число. В силу доказанного свойства 3 5 >0
, (0,00201) 2 >0
и .
Достаточно очевидно, что для любого натурального n
при a=0
степень a n
есть нуль. Действительно, 0 n =0·0·…·0=0
. К примеру, 0 3 =0
и 0 762 =0
.
Переходим к отрицательным основаниям степени.
Начнем со случая, когда показатель степени является четным числом, обозначим его как 2·m
, где m
— натуральное. Тогда . По каждое из произведений вида a·a
равно произведению модулей чисел a
и a
, значит, является положительным числом. Следовательно, положительным будет и произведение и степень a 2·m
. Приведем примеры: (−6) 4 >0
, (−2,2) 12 >0
и .
Наконец, когда основание степени a
является отрицательным числом, а показатель степени есть нечетное число 2·m−1
, то . Все произведения a·a
являются положительными числами, произведение этих положительных чисел также положительно, а его умножение на оставшееся отрицательное число a
дает в итоге отрицательное число. В силу этого свойства (−5) 3 .
Переходим к свойству сравнения степеней с одинаковыми натуральными показателями, которое имеет следующую формулировку: из двух степеней с одинаковыми натуральными показателями n
меньше та, основание которой меньше, а больше та, основание которой больше. Докажем его.
Неравенство a n свойств неравенств
справедливо и доказываемое неравенство вида a n (2,2) 7
и .
Осталось доказать последнее из перечисленных свойств степеней с натуральными показателями. Сформулируем его. Из двух степеней с натуральными показателями и одинаковыми положительными основаниями, меньшими единицы, больше та степень, показатель которой меньше; а из двух степеней с натуральными показателями и одинаковыми основаниями, большими единицы, больше та степень, показатель которой больше. Переходим к доказательству этого свойства.
Докажем, что при m>n
и 00
в силу исходного условия m>n
, откуда следует, что при 0
Осталось доказать вторую часть свойства. Докажем, что при m>n
и a>1
справедливо a m >a n
. Разность a m −a n
после вынесения a n
за скобки принимает вид a n ·(a m−n −1)
. Это произведение положительно, так как при a>1
степень a n
есть положительное число, и разность a m−n −1
есть положительное число, так как m−n>0
в силу начального условия, и при a>1
степень a m−n
больше единицы. Следовательно, a m −a n >0
и a m >a n
, что и требовалось доказать. Иллюстрацией этого свойства служит неравенство 3 7 >3 2
.
Свойства степеней с целыми показателями
Так как целые положительные числа есть натуральные числа, то все свойства степеней с целыми положительными показателями в точности совпадают со свойствами степеней с натуральными показателями, перечисленными и доказанными в предыдущем пункте.
Степень с целым отрицательным показателем , а также степень с нулевым показателем мы определяли так, чтобы оставались справедливыми все свойства степеней с натуральными показателями, выражаемые равенствами. Поэтому, все эти свойства справедливы и для нулевых показателей степени, и для отрицательных показателей, при этом, конечно, основания степеней отличны от нуля.
Итак, для любых действительных и отличных от нуля чисел a
и b
, а также любых целых чисел m
и n
справедливы следующие свойства степеней с целыми показателями :
a m ·a n =a m+n
;
a m:a n =a m−n
;
(a·b) n =a n ·b n
;
(a:b) n =a n:b n
;
(a m) n =a m·n
;
если n
– целое положительное число, a
и b
– положительные числа, причем ab −n
;
если m
и n
– целые числа, причем m>n
, то при 01
выполняется неравенство a m >a n
.
При a=0
степени a m
и a n
имеют смысл лишь когда и m
, и n
положительные целые числа, то есть, натуральные числа. Таким образом, только что записанные свойства также справедливы для случаев, когда a=0
, а числа m
и n
– целые положительные.
Доказать каждое из этих свойств не составляет труда, для этого достаточно использовать определения степени с натуральным и целым показателем, а также свойства действий с действительными числами. Для примера докажем, что свойство степени в степени выполняется как для целых положительных чисел, так и для целых неположительных чисел. Для этого нужно показать, что если p
есть нуль или натуральное число и q
есть нуль или натуральное число, то справедливы равенства (a p) q =a p·q
, (a −p) q =a (−p)·q
, (a p) −q =a p·(−q)
и (a −p) −q =a (−p)·(−q)
. Сделаем это.
Для положительных p
и q
равенство (a p) q =a p·q
доказано в предыдущем пункте. Если p=0
, то имеем (a 0) q =1 q =1
и a 0·q =a 0 =1
, откуда (a 0) q =a 0·q
. Аналогично, если q=0
, то (a p) 0 =1
и a p·0 =a 0 =1
, откуда (a p) 0 =a p·0
. Если же и p=0
и q=0
, то (a 0) 0 =1 0 =1
и a 0·0 =a 0 =1
, откуда (a 0) 0 =a 0·0
.
Теперь докажем, что (a −p) q =a (−p)·q
. По определению степени с целым отрицательным показателем , тогда . По свойству частного в степени имеем . Так как 1 p =1·1·…·1=1
и , то . Последнее выражение по определению является степенью вида a −(p·q)
, которую в силу правил умножения можно записать как a (−p)·q
.
Аналогично .
И .
По такому же принципу можно доказать все остальные свойства степени с целым показателем, записанные в виде равенств.
В предпоследнем из записанных свойств стоит остановиться на доказательстве неравенства a −n >b −n
, которое справедливо для любого целого отрицательного −n
и любых положительных a
и b
, для которых выполняется условие a. Так как по условию a0
. Произведение a n ·b n
тоже положительно как произведение положительных чисел a n
и b n
. Тогда полученная дробь положительна как частное положительных чисел b n −a n
и a n ·b n
. Следовательно, откуда a −n >b −n
, что и требовалось доказать.
Последнее свойство степеней с целыми показателями доказывается так же, как аналогичное свойство степеней с натуральными показателями.
Свойства степеней с рациональными показателями
Степень с дробным показателем мы определяли, распространяя на нее свойства степени с целым показателем. Иными словами, степени с дробными показателями обладают теми же свойствами, что и степени с целыми показателями. А именно:
Доказательство свойств степеней с дробными показателями базируется на определении степени с дробным показателем, на и на свойствах степени с целым показателем. Приведем доказательства.
По определению степени с дробным показателем и , тогда . Свойства арифметического корня позволяют нам записать следующие равенства . Дальше, используя свойство степени с целым показателем, получаем , откуда по определению степени с дробным показателем имеем , а показатель полученной степени можно преобразовать так: . На этом доказательство завершено.
Абсолютно аналогично доказывается второе свойство степеней с дробными показателями:
По схожим принципам доказываются и остальные равенства:
Переходим к доказательству следующего свойства. Докажем, что для любых положительных a
и b
, ab p
. Запишем рациональное число p
как m/n
, где m
– целое число, а n
– натуральное. Условиям p0
в этом случае будут эквивалентны условия m0
соответственно. При m>0
и a
Аналогично, при mb m
, откуда , то есть, и a p >b p
.
Осталось доказать последнее из перечисленных свойств. Докажем, что для рациональных чисел p
и q
, p>q при 00
– неравенство a p >a q
. Мы всегда можем привести к общему знаменателю рациональные числа p
и q
, пусть при этом мы получим обыкновенные дроби и , где m 1
и m 2
– целые числа, а n
— натуральное. При этом условию p>q
будет соответствовать условие m 1 >m 2
, что следует из . Тогда по свойству сравнения степеней с одинаковыми основаниями и натуральными показателями при 01
– неравенство a m 1 >a m 2
. Эти неравенства по свойствам корней можно переписать соответственно как и . А определение степени с рациональным показателем позволяет перейти к неравенствам и соответственно. Отсюда делаем окончательный вывод: при p>q
и 00
– неравенство a p >a q
.
Свойства степеней с иррациональными показателями
Из того, как определяется степень с иррациональным показателем , можно заключить, что она обладает всеми свойствами степеней с рациональными показателями. Так для любых a>0
, b>0
и иррациональных чисел p
и q
справедливы следующие свойства степеней с иррациональными показателями :
a p ·a q =a p+q
;
a p:a q =a p−q
;
(a·b) p =a p ·b p
;
(a:b) p =a p:b p
;
(a p) q =a p·q
;
для любых положительных чисел a
и b
, a0
справедливо неравенство a p b p
;
для иррациональных чисел p
и q
, p>q
при 00
– неравенство a p >a q
.
Отсюда можно сделать вывод, что степени с любыми действительными показателями p
и q
при a>0
обладают этими же свойствами.
Колмогоров А.Н., Абрамов А.М., Дудницын Ю.П. и др. Алгебра и начала анализа: Учебник для 10 — 11 классов общеобразовательных учреждений.
Гусев В.А., Мордкович А.Г. Математика (пособие для поступающих в техникумы).
Предварительный просмотр:
МУНИЦИПАЛЬНОЕ БЮДЖЕТНОЕ ОБЩЕОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ
СРЕДНЯЯ ОБЩЕОБРАЗОВАТЕЛЬНАЯ ШКОЛА № 11
МУНИЦИПАЛЬНОГО ОБРАЗОВАНИЯ ГОРОД – КУРОРТ АНАПА
Номинация «Физико-математические науки (математика)»
План – конспект урока по теме:
7 класс
Разработала: Быкова Е.А., учитель математики высшей квалификационной категории
Анапа, 2013
Открытый урок по алгебре в 7-м классе на тему:
«Свойства степени с натуральным показателем»
Цели урока:
Образовательные:
– отработка умений систематизировать, обобщать знания о степени с натуральным показателем, закрепить и усовершенствовать навыки простейших преобразований выражений, содержащих степени с натуральным показателем.
Воспитательные:
– воспитание познавательной активности, чувства ответственности, культуры общения, культуры диалога.
Развивающие:
— развитие зрительной памяти, математически грамотной речи, логического мышления, сознательного восприятия учебного материала.
Задачи:
1.
Предметные:
повторить, обобщить и систематизировать знания по теме, создать условия контроля (взаимоконтроля) усвоения знаний и умений; продолжить формирование мотивации обучающихся к изучению предмета.
2.
Метапредметные:
развивать операционный стиль мышления, способствовать приобретению учащимися навыков общения при совместной работе,активизировать их творческое мышление; продолжить формирование определенных компетенций обучающихся, которые будут способствовать их эффективной социализации, навыков самообразования и самовоспитания
3.
Личностные:
воспитывать культуру, способствовать формированию личностных качеств, направленных на доброжелательное, толерантное отношение к людям, жизни; воспитывать инициативу и самостоятельность в деятельности; подвести к пониманию необходимости изучаемой темы для успешной подготовки к государственной итоговой аттестации.
Тип урока:
обобщающий урок по теме.
Вид урока:
комбинированный.
Структура урока:
1. Организационный момент.
2. Сообщение темы, целей и задач урока.
3. Воспроизведение изученного и его применение в стандартных ситуациях.
4. Перенос приобретенных знаний, их первичное применение в новых или изменённых условиях, с целью формирования умений.
5.Элементы здорорвьесберегающих технологий.
6.Самостоятельное выполнение учащимися заданий под контролем учителя.
7.Подведение итогов урока и постановка домашнего задания.
Оборудование:
мультимедийный проектор, компьютер.
Презентация в программе Microsoft Office Power Point 2007
(Приложение 1)
План урока:
Этап урока
Время
Организационный момент.
Нацелить учащихся на урок
1 мин.
Проверка домашнего задания
Коррекция ошибок
3 мин.
Сообщение темы, целей и задач урока.
Постановка целей урока
1 мин.
Устная работа. Повторение свойств степени с натуральным показателем.
Актуализировать опорные знания
7 мин.
Тренировочные упражнения.
Сформировать навык преобразования степеней с натуральным показателем.
10 мин.
Физкультурная пауза.
Применение здоровья сберегающих технологий
2 мин.
Индивидуальная проверочная работа по карточкам.
Коррекция ошибок
12 мин
Итоги урока.
Обобщить теоретические сведения, полученные на уроке
2 мин
Постановка домашнего задания.
Разъяснить содержание домашнего задания
2 мин
Литература:
1. Алгебра: учебн. для 7 кл. общеобразоват. учреждений / Ю.Н. Макарычев, Н.Г.Миндюк и др.; под редакцией С.А. Теляковского. – М.: Просвещение, 2008.
2. Звавич Л.И., Кузнецова Л.В., Суворова С.Б. Дидактические материалы по алгебре для 7 класса. – М.: Просвещение, 2009.
3. Сборник тестовых заданий для тематического и итогового контроля. Алгебра 7 класс./ С.А. Пушкин, И.Л. Гусева. – М.: «Интеллект», 2013.
а) Повторение свойств степени с натуральным показателем. Дана таблица. В левом столбце заполнить пропущенные места, в правом – выполнить задания.
Степенью числа
а
с натуральным показателем
п
называется ____________
п ____________,
каждый из которых равен
а.
1. Представьте в виде степени произведение:
а). (-8)
*
(-8)
*
(-8)
*
(-8)
*
(-8)
*
;
б). (x-y)
*
(x-y)
*
(x-y)
*
(x-y)
*
;
2. Возведите в степень:
3
4
; (-0,2)
3
; (2/3)
2
Назовите основание и показатель записанных степеней.
При умножении степеней с одинаковыми основаниями ___________ оставляют прежним, а ___________ складывают.
Выполните действия:
а
4
*
а
12
;
а
6
*
а
9
*
а
;
3
2
*
3
3
При делении степеней с одинаковыми основаниями ___________ оставляют прежним, а из __________ числителя _________ __________ знаменателя.
Выполните действия:
а
12
:
а
4
;
п
9
:
п
3
:
п
;
3
5
:
3
2
При возведении степени в степень _______________ оставляют прежним, а __________ перемножают.
Выполните действия:
;
(m
3
)
7
; (k
4
)
5
; (4
2
)
3
При возведении в степень произведения возводят в эту степень _____________ ____________ и результаты перемножают.
Выполнить возведение в степень:
(-2 a
3
b
2
)
5
; (1/3p
2
q
3
)
3
Степень числа
a
, не равного нулю, с нулевым показателем равна
Вычислите:
3x
0
при x= 2,6
б) Выполняя задания на преобразования выражений, содержащих степени, ученик допустил следующие ошибки:
(запись на доске)
1) а)
; б)
;
в)
; г)
;
2) а)
; б)
;
в)
; г)
;
3) а)
; б)
;
в)
.
Какие определения, свойства, правила не знает ученик?
5.
Тренировочные упражнения.
№ 447 – на доске и в тетрадях с подробным комментированием, используя свойства степеней;
№ 450 (а,в) – на доске и в тетрадях;
№ 445 – устно.
6. Физминутка
Быстро встали, улыбнулись,
Выше-выше подтянулись.
Ну-ка плечи распрямите,
Поднимите, опустите.
Вправо, влево повернитесь,
Рук коленями коснитесь.
Сели, встали, сели, встали,
И на месте побежали.
Учится с тобою молодёжь
Развивать и волю, и смекалку.
7. Индивидуальная проверочная работа.
Каждый учащийся выполняет задания, к ним прилагается ключ, в котором использован весь алфавит, чтобы исключить угадывание ответов по буквам. В случае правильного решения – правильное слово.
Задания для каждого ряда индивидуальные.
№ п/п
Задание 1 ряд
№ п/п
Задание 2 ряд
№ п/п
Задание 3 ряд
m
3
*
m
2
*
m
8
a
4
*
a
3
*
a
2
a
4
*
a
*
a
3
*
a
p
20
: p
17
(2
4
)
5
: (2
7
)
2
(7x)
2
c
5
: c
0
3
*
3
2
*
3
0
p
*
p
2
*
p
0
(3a)
3
(2y)
5
c
*
c
3
*
c
m
*
m
5
*
m
3
*
m
0
(m
2
)
4
*
m
m
*
m
4
*
(m
2
)
2
*
m
0
2
14
: 2
8
(2
3
)
2
(2
3
)
7
: (2
5
)
3
(-x)
3
*
x
4
(-x
3
)
*(-
x)
4
X
3
*
(-x)
4
(p
*
p
3
) : p
5
(p
2
*
p
5
) : p
4
*
p
0
(p
2
)
4
: p
5
3
7
*
(3
2
)
3
: 3
10
(3
5
)
2
*
3
7
: 3
14
(3
4
)
2
*
(3
2
)
3
: 3
11
Ключ
32y
5
49x
2
27a
3
m
13
81a
3
16a
4
10y
5
9y
7
32x
5
49y
3
Результаты работы высвечиваются на слайде для самопроверки:
Математика
8. Итоги урока:
Подведение итогов урока, выставление оценок.
– Перечислите свойства степени с натуральным показателем.
Оценки за урок поставим после проверки работы с тестами, учитывая, ответы тех учащихся, которые отвечали в течение урока.
Отгадайте кроссворд
По вертикали:
Он делит делимое
Элементарная фигура на плоскости
Верное равенство
Единица с девятью нулями
Его складывают с подобным
Два в степени три
По горизонтали:
2. Число сторон в треугольнике
4. Сумма одночленов
5. Суммировать
7. Отрезок, соединяющий точку окружности с её центром
8. Имеет числитель и знаменатель
9. Задание на дом:
Степенью числа а с натуральным показателем п называется ____________ п ____________, каждый из которых равен а. 1. Представьте в виде степени произведение: а). (-8) * (-8) * (-8) * (-8) * (-8) * ; б). (x-y)* (x-y) * (x-y) * (x-y) * ; 2. Возведите в степень: 3 4 ; (-0,2) 3 ; (2 /3) 2 Назовите основание и показатель записанных степеней. При умножении степеней с одинаковыми основаниями ___________ оставляют прежним, а ___________ складывают. Выполните действия: а 4 * а 12 ; а 6 * а 9 * а; 3 2 * 3 3 При делении степеней с одинаковыми основаниями ___________ оставляют прежним, а из __________ числителя _________ __________ знаменателя. Выполните действия: а 12: а 4 ; п 9: п 3: п; 3 5: 3 2 При возведении степени в степень _______________ оставляют прежним, а __________ перемножают. Выполните действия: ; (m 3) 7 ; (k 4) 5 ; (4 2) 3 При возведении в степень произведения возводят в эту степень _____________ ____________ и результаты перемножают. Выполнить возведение в степень: (-2 a 3 b 2) 5 ; (1 /3p 2 q 3) 3 Степень числа a , не равного нулю, с нулевым показателем равна Вычислите: 3 x 0 при x = 2,6 Повторим!
Мозговой штурм
Быстро встали, улыбнулись, Выше-выше подтянулись. Ну-ка плечи распрямите, Поднимите, опустите. Вправо, влево повернитесь, Рук коленями коснитесь. Сели, встали, сели, встали, И на месте побежали. Учится с тобою молодёжь Развивать и волю, и смекалку.
Индивидуальная проверочная работа № п / п Задание 1 ряд № п/п Задание 2 ряд № п/п Задание 3 ряд 1 m 3 * m 2 * m 8 1 a 4 * a 3 * a 2 1 a 4 * a * a 3 * a 2 p 20: p 17 2 (2 4) 5: (2 7) 2 2 (7x) 2 3 c 5: c 0 3 3 * 3 2 * 3 0 3 p * p 2 * p 0 4 (3a) 3 4 (2y) 5 4 c * c 3 * c 5 m * m 5 * m 3 * m 0 5 (m 2) 4 * m 5 m * m 4 * (m 2) 2 * m 0 6 2 14: 2 8 6 (2 3) 2 6 (2 3) 7: (2 5) 3 7 (-x) 3 * x 4 7 (-x 3) *(- x) 4 7 -x 3 * (-x) 4 8 (p * p 3) : p 5 8 (p 2 * p 5) : p 4 * p 0 8 (p 2) 4: p 5 9 3 7 * (3 2) 3: 3 10 9 (3 5) 2 * 3 7: 3 14 9 (3 4) 2 * (3 2) 3: 3 11
Проверь себя! Ключ! А Б В Г Д Е Ж З И К m 9 32y 5 81 a 9 x 3 49x 2 m 5 p 4 c 5 27a 3 Л М Н О П Р С Т У Ф 64 3 4 p 3 27 2 5 x 7 p 6 m 3 m 13 a 8 Х Ц Ч Ш Щ Ъ Ь Ы Э Ю 81a 3 c 7 16a 4 25 10y 5 9y 7 -x 7 a 2 32x 5 49y 3 Я x 5
математика
ОТГАДАЙТЕ КРОССВОРД По вертикали: 1. Он делит делимое 2. Элементарная фигура на плоскости 3. Верное равенство 4. Единица с девятью нулями 5. Его складывают с подобным 6. Два в степени три По горизонтали: 2. Число сторон в треугольнике 4. Сумма одночленов 5. Суммировать 7. Отрезок, соединяющий точку окружности с её центром 8. Имеет числитель и знаменатель
Итог урока Выставление оценок Задание на дом Ответить на вопросы стр. 101, № 450(б,г) , № 534, № 453.
основание и показатель степени. Онлайн калькулятор
Степень числа — это выражение, обозначающее краткую запись произведения одинаковых сомножителей.
Рассмотрим умножение одинаковых чисел, например:
5 · 5 · 5 = 125.
Произведение 5 · 5 · 5 можно записать так: 53 (пять в третьей степени). Выражение 53 — это степень. Следовательно,
5 · 5 · 5 = 53 = 125.
Рассмотрим выражение 53 . В этом выражении число 5 — основание степени, а число 3 — показатель степени.
Основание степени — это повторяющийся множитель. Показатель степени — это число, указывающее количество повторений, то есть показатель степени показывает сколько одинаковых множителей содержится в произведении.
Читаются степени так:
72 — семь во второй степени.
Вторую степень числа также называют квадратом этого числа. Следовательно, выражение 72 можно прочесть так: семь в квадрате или квадрат числа семь.
23 — два в третьей степени.
Третью степень числа также называют кубом этого числа. Следовательно, выражение 23 можно прочесть так: два в кубе или два куб.
64 — шесть в четвёртой степени.
1015 — десять в пятнадцатой степени.
an — a в энной степени или a в степени эн.
Пример. Записать в виде степени:
a) 5 · 5;
б) 10 · 10 · 10 · 10;
в) 8 · 8 · 8.
Решение:
a) 5 · 5 = 52;
б) 10 · 10 · 10 · 10 = 104;
в) 8 · 8 · 8 = 83.
Возведение в степень
Возведение числа в степень — это вычисление произведения одинаковых множителей. Например, возвести число 2 в третью степень (23) — это значит найти произведение 2 · 2 · 2 , то есть
23 = 2 · 2 · 2 = 8.
Результат возведения в степень называется степенью (также как и само выражение, значение которого вычисляется). В выражении:
23 = 8,
2 — это основание степени, 3 — показатель степени, 8 — степень.
Пример. Вычислите:
a) 112;
б) 25;
в) 104.
Решение:
a) 112 = 11 · 11 = 121;
б) 25 = 2 · 2 · 2 · 2 · 2 = 32;
в) 104 = 10 · 10 · 10 · 10 = 10000.
Выражения со степенями. Порядок действий
Если выражение не содержит скобки и содержит степени, то сначала выполняется возведение в степень в порядке следования степеней (слева направо), а затем все остальные арифметические действия. Если выражение содержит скобки, то сначала выполняются действия в скобках, с учётом всех правил порядка выполнения действий.
Рассмотрим два выражения:
52 + 22
и
(5 + 2)2
В соответствии с порядком выполнения действий в первом случае сначала выполняется возведение в степень, а затем вычисляется сумма. Во втором случае сначала вычисляется сумма, а затем результат возводится в квадрат.
52 + 22 = 25 + 4 = 29,
(5 + 2)2 = 72 = 49.
Пример 1. Найти значение выражения:
5 · (10 — 8)3.
Решение: Сначала выполняется действие, заключённое в скобки:
1) 10 — 8 = 2.
Затем, по правилам порядка действий, выполняется возведение в степень:
2) 23 = 2 · 2 · 2 = 8.
И последним действием вычисляется произведение:
3) 5 · 8 = 40.
Ответ: 5 · (10 — 8)3 = 40.
Пример 2. Вычислить:
a) (4 + 2) · 32;
б) 3 · 52 — 50;
в) 3 · 4 + 62.
Решение:
a) (4 + 2) · 32 = 54
4 + 2 = 6
32 = 9
6 · 9 = 54
б) 3 · 52 — 50 = 25
52 = 25
3 · 25 = 75
75 — 50 = 25
в) 3 · 4 + 62 = 48
62 = 36
3 · 4 = 12
12 + 36 = 48
Калькулятор возведения в степень
Данный калькулятор поможет вам выполнить возведение в степень. Просто введите основание с показателем степени и нажмите кнопку Вычислить.
Свойства степени с натуральными показателями
Однажды к учителю подошел ученик поймавший бабочку и спросил: «Учитель, какая у меня в руках бабочка: живая или мертвая?». Учитель, даже не взглянув на ученика, ответил: «Все в твоих руках».
Вот и наш сегодняшний урок в наших руках.
В Древней Индии была такая легенда. Стоит камень размером в кубический километр, в миллион раз тверже алмаза. Один раз в миллион лет к нему прилетает птичка и трется клювом о камень. В конце концов в результате этого камень износится. Как вы думаете, сколько лет понадобится для того, чтобы камень износился до основания?
Вычисления математиков показывают, что для этого понадобится1035лет.
Свойства степени с натуральным показателем
урок по алгебре, 7 класс
Цель:
1. вывести свойства степени с натуральными показателями;
2. изучить свойства степени с натуральным показателем, их формулировки и символическую запись;
3. выработать практические умения и навыки по применению изученных свойств.
Большая часть математических утверждений проходит в своем становлении три этапа.
На первом этапе человек в ряде конкретных случаев подмечает одну и ту же закономерность.
На втором этапе он пытается сформулировать подмеченную закономерность в общем виде, т.е. предполагает, что эта закономерность действует не только в рассмотренных случаях, но и во всех других аналогичных случаях.
На третьем этапе он пытается доказать, что закономерность, сформулированная в общем виде, на самом деле верна.
Открытие первое
Открытие второе
Открытие третье
Запомните:
Правило 1.При умножении степеней с одинаковыми основаниями показатели складываются, а основание остается неизменным.
Правило 2.При делении степеней с одинаковыми основаниями показатели вычитаются, а основание остается неизменным.
Правило 3.При возведении степени в степень
показатели перемножаются, а основание остается неизменным.
Свойства степеней
Ответы
Найдите ошибки
210
54
220
7
8х3
1
а6
210
Подумайте, чем можно заменить ?
Ответы
n10
x12
a0
b14
k55
m15
d30
c45
t10
54
24
77
4
4
0
Высказывания Козьмы Пруткова
Выполните преобразования. Используя найденные ответы, запишите в таблицах два высказывания Козьмы Пруткова:
Заполните свободные клетки квадрата так, чтобы произведение выражений каждого столбца, каждой строки и диагонали равнялосьх12
Список использованной литературы:
1. Лебединцева Е.А., Беленкова Е.Ю. Алгебра 7 класс. Задание для обучения и развития учащихся. – М.: Интеллект-центр, 2005.
2. Мордкович А.Г. и др. Алгебра 7 класс, Учебник, М.: Мнемозина, 2007;
3. Мордкович А.Г. и др. Алгебра 7 класс, Задачник, М.: Мнемозина, 2007;
4. Мордкович А.Г., Тульчинская Е.Е. Алгебра: Тесты для 7-9 класса общеобразовательных учреждений. – М.: Мнемозина, 2002.
Индикаторы природных кислот и щелочей
Есть много обычных товаров для дома и садовых растений, которые можно использовать в качестве индикаторов pH. Большинство растений содержат антоцианы, чувствительные к pH, что делает их идеальными для определения кислотного и щелочного уровней. Многие из этих естественных индикаторов pH имеют широкий диапазон цветов.
Растения, которые можно использовать для проверки уровня pH
Мир природы дал нам множество растений, от свеклы до винограда и лука, которые можно использовать для проверки уровня pH раствора.Эти естественные индикаторы pH включают:
Свекла: Очень щелочной раствор (с высоким pH) изменит цвет свеклы или свекольного сока с красного на фиолетовый.
Ежевика: Ежевика, черная смородина и черная малина меняют цвет с красного в кислой среде на синий или фиолетовый в основной среде.
Черника: Черника голубая при pH 2,8–3,2, но становится красной по мере того, как раствор становится еще более кислым.
Вишни: Вишни и их сок красны в кислом растворе, но они становятся синими или пурпурными в основном растворе.
Порошок карри: Карри содержит пигмент куркумин, цвет которого изменяется от желтого при pH 7,4 до красного при pH 8,6.
Лепестки дельфиниума: Антоциановый дельфинидин изменяется от голубовато-красного в кислом растворе до фиолетово-синего в основном растворе.
Лепестки герани: Герань содержит антоцианин пеларгонидин, цвет которого изменяется от оранжево-красного в кислом растворе до синего в основном растворе.
Виноград: Красный и фиолетовый виноград содержат множество антоцианов.Синий виноград содержит моноглюкозид мальвидина, цвет которого изменяется от темно-красного в кислом растворе до фиолетового в основном растворе.
Листья конского каштана: Замочите листья конского каштана в спирте для экстракции флуоресцентного красителя эскулина. Эскулин бесцветен при pH 1,5, но становится флуоресцентно-синим при pH 2. Для наилучшего эффекта светите черным светом на индикаторе.
Утренняя слава: Утренняя слава содержит пигмент, известный как «небесно-синий антоциан», который меняется с пурпурно-красного на пурпурный при pH 6.От 6 до синего при pH 7,7.
Лук: Лук является обонятельным индикатором. Вы не чувствуете запаха лука в сильно щелочных растворах. Красный лук также меняет цвет с бледно-красного в кислом растворе на зеленый в основном растворе.
Petunia Petals: Антоциановый петунин изменяется от красновато-пурпурного в кислом растворе до фиолетового в основном растворе.
Ядовитая примула: Primula sinensis имеет оранжевые или синие цветы. Цветки апельсина содержат смесь пеларгонинов.В синих цветках содержится мальвин, цвет которого меняется от красного к пурпурному по мере того, как раствор переходит от кислого к щелочному.
Пурпурные пионы: Пеонин меняется от красновато-пурпурного или пурпурного в кислом растворе до темно-фиолетового в основном растворе.
Красная (пурпурная) капуста: Краснокочанная капуста содержит смесь пигментов, используемых для обозначения широкого диапазона pH.
Лепестки роз: Оксониевая соль цианина меняет цвет с красного на синий в щелочном растворе.
Куркума: Эта специя содержит желтый пигмент куркумин, который меняет свой цвет с желтого на pH 7.4 становится красным при pH 8,6.
Если у вас нет под рукой какого-либо из вышеперечисленных материалов, вы также можете использовать некоторые обычные бытовые химикаты для проверки уровня pH. Это включает:
Пищевая сода: Пищевая сода будет шипеть при добавлении ее в кислый раствор, например уксус, но не будет шипеть в щелочном растворе. Реакция не всегда полностью меняется, поэтому, хотя пищевую соду можно использовать для проверки раствора, ее нельзя использовать повторно.
Помада, меняющая цвет: Вам нужно будет протестировать свою изменяющую цвет помаду, чтобы определить ее диапазон pH, но большинство косметических средств, которые меняют цвет, реагируют на изменения pH (они отличаются от косметики, которая меняет цвет в зависимости от угла свет).
ExLax Tablets: Эти таблетки содержат фенолфталеин, который является индикатором pH, который бесцветен в растворах с более кислотным, чем pH 8,3, и от розового до темно-красного цвета в растворах с более щелочным, чем pH 9.
Экстракт ванили: Экстракт ванили является обонятельным индикатором.Вы не можете почувствовать характерный запах при высоких значениях pH, потому что молекула находится в ионной форме.
Сода для стирки: Как и пищевая сода, сода шипит в кислотном растворе, но не в щелочном растворе.
индикаторов — Chemistry LibreTexts
Цели обучения
Объясните изменение цвета индикаторов.
Определите константы кислотной диссоциации K a или K ai индикаторов.
Индикаторы — это вещества, растворы которых меняют цвет из-за изменения pH. Это так называемые кислотно-основные индикаторы. Обычно это слабые кислоты или основания, но их сопряженные основания или кислотные формы имеют разный цвет из-за различий в их спектрах поглощения. Знаете ли вы, что цвет цветков гортензии зависит от pH почвы, в которой они выращены?
Рисунок \ (\ PageIndex {1} \): На этом рисунке показаны различные цвета цветов гортензии. (CC BY-SA 4.-} \) преобладает цвет, тогда как цвет из-за \ (\ mathrm {\ color {Blue} HIn} \) доминирует, если \ (\ ce {[H +]} <\ dfrac {K _ {\ large \ textrm {ai}} } {10} \). Вышеприведенное уравнение показывает, что изменение цвета является наиболее чувствительным, когда \ (\ ce {[H +]} = K _ {\ large \ textrm {ai}} \) в числовом значении.
Мы определяем p K ai = — log ( K ai ), а значение p K ai также является значением pH, при котором цвет индикатора наиболее чувствителен к изменениям pH. .-}] = [\ mathrm {\ color {Blue} HIn}] \). Другими словами, когда pH такой же, как p K ai , существуют равные количества кислотной и основной форм. Когда две формы имеют одинаковую концентрацию, изменение цвета наиболее заметно.
Цвета субстанций делают мир прекрасным местом. Благодаря цветам и структуре цветы, растения, животные и минералы проявляют свой уникальный характер. Многие индикаторы извлекаются из растений. Например, краснокочанный сок и чайные пигменты имеют разные цвета при разном pH.Цвет чая темнеет в базовом растворе, но цвет становится светлее при добавлении лимонного сока. Сок краснокочанной капусты становится синим в щелочном растворе, но имеет отчетливый красный цвет в кислом растворе.
Общие индикаторы: некоторые общие индикаторы и их значения pK ai (также называемые pK a ) представлены в виде таблицы.
Фенолфталеин в приведенной выше таблице имеет значение p K ai , равное 9,7, что является наиболее близким к значению pH точки эквивалентности при этом титровании. Этот индикатор бесцветен в кислом растворе, но светлый РОЗОВЫЙ появляется, когда pH> 8. Цвет становится более ИНТЕНСИВНЫМ РОЗОВЫМ по мере повышения pH. Парад интенсивностей цвета показан ниже:
Точка эквивалентности — это когда цвет меняется наиболее быстро, а не когда раствор изменил цвет.Неправильное использование индикаторов приведет к неточности результатов титрования.
Цвета индикаторного раствора
Индикаторы постепенно меняют цвет при различных значениях pH. Предположим, что кислотная форма имеет синий цвет, а основная форма — красный цвет. Ниже показано изменение цвета при разном pH. Цвет фона влияет на их внешний вид и наше восприятие.
Длинный растянутый цвет в середине последней строки имеет одинаковую интенсивность СИНИЙ и КРАСНЫЙ.Если цвет раствора соответствует этому, pH будет таким же, как p K ai индикатора, при условии, что сопряженные формы индикатора имеют СИНИЙ и КРАСНЫЙ цвета.
вопросов
Растения содержат множество природных индикаторов. Краситель краснокочанной капусты, фиолетовый цвет винограда и даже цвет некоторых цветов — вот некоторые примеры. Почему некоторые фрукты меняют цвет при созревании?
Выберите верное утверждение:
Все слабые кислоты являются индикаторами.
Все слабые базы являются индикаторами.
Слабые кислоты и основания являются индикаторами.
Все индикаторы — слабые кислоты.
Пара кислотно-основных конъюгатов имеет разные цвета.
Любой индикатор меняет цвет при pH его раствора 7.
Все индикаторы меняют цвет при pH 7 (да / нет)?
Решения
Ответ \ (\ ce {[H +]} \) сока меняется. Подсказка … Изменения pH или \ (\ ce {[H +]} \) вызывают изменение цвета красителя, если их сопряженные пары кислота-основание имеют разные цвета. Могут быть и другие причины. Указывают ли цвета, насколько они хороши или плохи на вкус?
Ответ д. Подсказка … Изменение цвета является обязательным условием для индикаторов.
Ответ Нет! Совет … Фенолфталеин меняет цвет при pH ~ 9. Бромтимоловый синий имеет значение p K n , равное 7.1. При pH 7 его цвет меняется с желтого на синий. Некоторые индикаторы меняют цвет при pH, отличном от 7.
Авторы и ссылки
Натуральных экстрактов растений как кислотно-основных индикаторов и определение их значения pKa
Обычно используемые индикаторы для кислотно-основного титрования являются синтетическими, и эта работа была направлена на выявление экологически чистых природных индикаторов и определение их значений pKa. Аналитический потенциал экстрактов цветов очень многообещающий, что видно из их применения в кислотно-щелочной титриметрии.Было обнаружено, что эти отобранные цветочные экстракты лучше подходят для титрования сильного кислотно-сильного основания, чем слабого кислотно-сильного основания. Мы получили резкое и четкое изменение цвета от красного до коричневато-желтого для экстракта Bougainvillea glabra , от красного до желтого для экстракта Bauhinia purpurea и от красного до коричневато-желтого для экстракта Impatiens balsamina . Все три экстракта цветов дали отчетливое изменение цвета с помощью кислот и оснований, и изменение цвета поддерживалось с помощью различных кислот и оснований.Резкий контраст между их цветами в кислоте и основе сделал пигмент подходящим для использования в качестве кислотно-основных индикаторов. Поскольку эти цветочные экстракты имеют очень простую, экономичную, экологически безопасную процедуру экстракции и отличные характеристики с резким изменением цвета в конечных точках титрования, можно было бы заменить стандартные индикаторы, используемые в обычных лабораториях, индикаторами естественных цветов.
1. Введение
Титрование — это наиболее распространенный лабораторный метод количественного химического анализа, который используется для определения концентрации анализируемого вещества.Большинство современных лабораторий оснащены цифровыми автоматическими титраторами, которые оснащены датчиками (датчик pH / электрод напряжения), некоторые из них не требуют индикаторов, точность высока, а количество ошибок, связанных с человеческим фактором, также меньше, чем при использовании традиционных методов титрования. Однако кинетические факторы, касающиеся химической реакции и реакции индикаторной системы, имеют первостепенное значение. Конфигурация ячейки, перемешивание и расположение детектора конечной точки и ввода титранта должны быть рассмотрены для обеспечения высокой точности.Поршневые бюретки и перистальтические насосы обычно используются в качестве устройств для автоматической перекачки титранта в автоматические титраторы. Поршневые бюретки очень надежны, но дороги, в то время как перистальтические насосы очень универсальны, но требуют частой калибровки из-за постоянных изменений физических свойств используемых гибких трубок и имеют относительно короткий срок службы [1]. Стоимость автоматических титраторов вместе с недостатками, рассматриваемыми как основные препятствия для использования автоматических титраторов во многих развивающихся странах мира, и, следовательно, традиционные титриметрические методы все еще широко используются в аналитических и исследовательских лабораториях этих стран.
С тех пор, как мир стал осведомлен об экологических проблемах, различные части растений, такие как цветы и листья, являются символическими и считаются символом любовных желаний. Таким образом, цветы — это чудо природы. Синтетические соединения сильно загрязняют окружающую среду, вредны, опасны и намного дороже как для исследовательской, так и для аналитической работы. Поэтому многие ученые всего мира активно проводят различные исследования в этой области натуральных продуктов, поскольку они менее опасны, дешевы, легкодоступны и экологичны [2].
Химические вещества обладают очевидным изменением цвета анализируемого вещества и реакционной смеси титранта, очень близко к точке текущего титрования, известной как индикатор, который помогает исследовать и определять точку эквивалентности при кислотно-основном титровании [3, 4] . Природные красители и пигменты растений — это сильно окрашенные вещества, которые могут менять цвет при изменении pH [5]. Цвета частей растений выражают их неповторимый характер. Некоторые органические и неорганические соединения ответственны за свойство окраски частей растения, такие как флавоноиды, флавонолы, ацилированные флавоноиды, антоцианы, глюкозилированный ацилированный антоцианин, хинины, имины, полиметины, нафтахиноны, антрахметиноноиды, индигидропиригоиды, дигидропиригоиды.[6]. Некоторые из этих соединений показывают разные цвета при разном pH, и, таким образом, это свойство можно использовать в качестве естественного индикатора. Индикатор pH — это просто слабое кислотно-слабое основание с разноокрашенными кислотными и сопряженными формами основания. Синие и красные пигменты цветов были выделены и всесторонне изучены Вильштеттером в 1913 году. Были разработаны природные индикаторы, такие как лакмусовая бумажка, для обозначения определенных уровней pH. Вещества в растительных продуктах, таких как чай, краснокочанная капуста или виноград, вступают в реакцию с кислотами или основаниями, что приводит к изменениям на молекулярном уровне, что приводит к различию их цвета при разных значениях pH.Сок краснокочанной капусты использовался как естественный индикатор pH [7]. Этот индикатор содержит антоцианин, пигмент которого по-разному реагирует на кислоты и основания [7].
Каждое соединение, которое может действовать как индикатор, имеет специфическое значение p K и , и это важный физический параметр, указывающий на кислотность молекул. Для большинства индикаторов диапазон pH находится в пределах ± 1 от значения p K до . Целью данной работы было выявление экологически чистых природных индикаторов и определение их значений pKa.Для этого исследования использовали водные экстракты трех растений Bougainvillea glabra, Impatiens balsamina, и Bauhinia purpurea .
Бугенвиллея глабра — вечнозеленый плетистый кустарник с колючими стеблями. Крошечные белые цветы обычно появляются группами, окруженными разноцветными бумажными прицветниками. Эти прицветники содержат одиннадцать типов пигментов бугенвиллей-v (бетацианин) [8].
Impatiens balsamina (садовый бальзам) произрастает в таких местах Южной Азии, как Индия, Шри-Ланка, Бангладеш и Мьянма.Это однолетнее растение высотой 20–75 см с толстым, но мягким стеблем. Листья расположены по спирали. Цветки розовые, красные, лиловые, лиловые или белые. Лепестки содержат антоцианидин и пеларгонидин в качестве красящих пигментов [9].
Bauhinia purpurea , пурпурная орхидея, — экзотическое тропическое дерево, которое цветет в течение длительного периода времени. Красивые и похожие на орхидеи цветы Bauhinia purpurea произрастают в Индии. Лепестки Bauhinia содержат в качестве красящих пигментов халкон и бутеин [10].
2. Материалы и методы
2.1. Приготовление экстрактов
Цветки зеленых растений Bougainvillea glabra и Impatiens balsamina были получены из сада Восточного университета, Ченкалади, Баттикалоа, Шри-Ланка, а цветок Bauhinia purpurea был получен из Thambiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri, Ampiluvilri. . Лепестки каждого цветка собирали, промывали дистиллированной водой и зажимали между подушечками впитывающей бумаги для удаления воды с поверхности.Свежие лепестки (5 г) каждого цветка переносили в химические стаканы (100 мл), содержащие 50 мл дистиллированной воды отдельно. Его нагревали до 50–60 ° С в течение 10 минут. Экстракты трех цветков разделяли фильтрованием через чистую фильтровальную бумагу Whatman® № 1 (размер пор 11 мкм мкм). Экстракты хранили в сухом и темном месте.
2.2. Кислотно-основное титрование
Три типа титрования, такие как сильное кислотно-сильное основание, сильное кислотно-слабое основание и слабое кислотно-сильное основание, были выполнены с использованием экстрактов цветов в качестве индикаторов, и их точность сравнивалась с коммерчески доступными синтетическими индикаторами, такими как как метиловый оранжевый, метиловый красный и фенолфталеин.Каждое титрование проводили в трех экземплярах.
2.3. Приготовление растворов с различным pH
Мерные колбы (50 мл) были помечены номерами от 1 до 10. В каждую колбу добавляли экстракт цветов (2,0 мл). Растворы 0,1 M Na 2 HPO 4 и 0,1 M KH 2 PO 4 также добавляли в каждую колбу, как указано в таблице 1. Каждый раствор разбавляли до метки дистиллированной водой. Эту процедуру повторили для всех трех экстрактов цветов [11].
Колба No.
KH 2 PO 4 (мл)
Na 2 HPO 4 (мл)
1
10,0
0,0
0,0
0,0 902
1,0
3
10,0
2,0
4
20,0
10,0
5
10.0
10,0
6
10,0
20,0
7
2,0
10,0
8
2,0
20,0 902
902 902 902
902 902 902 902 902 902 902 902
10
0,0
10,0
2,4. Измерение pH растворов
pH каждого раствора измеряли с помощью зондов pH (PHC301), подключенных к откалиброванному многопараметрическому датчику Hach hq40d.
Калибровка прибора: (I) зонд pH был подсоединен к прибору, и контргайка была установлена правильно. Измеритель был включен. (II) После нажатия кнопки «калибровка» зонд был промыт деионизированной водой, и его осторожно, без пузырьков воздуха, поместили в стандартный буферный раствор pH при легком перемешивании. (III) Затем значок « чтение ». (IV) Та же процедура (III) была проделана для каждого стандартного раствора pH. (V) Наконец, была нажата кнопка« Готово », и калиброванные данные были сохранены.
2,5. Спектрофотометрическое определение значений pKa индикаторов
Цветочный экстракт (2,0 мл), дистиллированная вода (10,0 мл) и 8 капель кон. HCl переносили в 50 мл мерную колбу, обозначенную как A , и раствор разбавляли до метки дистиллированной водой. Во вторую мерную колбу, обозначенную как B , переносили экстракт цветков (2,0 мл), дистиллированную воду (10,0 мл) и 24 капли 4 М NaOH, и раствор доводили до метки дистиллированной водой.Это повторили для всех трех цветочных экстрактов.
Для этого исследования использовался спектрофотометр Biobase BK-D580. УФ-видимые спектры были получены между 300 и 800 нм для A и B каждого раствора экстракта цветов и для метилового красного. Была выбрана одна длина волны ( λ, 1 ), при которой раствор A поглощает сильно, а раствор B — слабо, и вторая длина волны ( λ 2 ), при которой раствор A поглощает слабо, а раствор B — сильно.Наконец, абсорбция каждого набора из 10 растворов была сведена в таблицу относительно холостого раствора при выбранных длинах волн λ 1 и λ 2 (Таблица 2). Здесь холостой раствор включает все химические вещества и дистиллированную воду, кроме экстракта цветков [11–14].
Вышеупомянутая процедура была проделана для всех трех экстрактов цветов, и результаты их экспериментов сравнивались с показателем метилового апельсина.
Индикаторы представляют собой слабое основание или слабую кислоту, которые имеют разные цвета при разном pH. Равновесная реакция индикатора показана в следующем уравнении [12]:
Большинство индикаторов представлены в виде HIn в растворе сильной кислоты и имеют соответствующий цвет HIn, тогда как в растворе сильного основания большинство индикаторов представлены в виде In — и имеют соответствующий цвет In — .
Равновесное выражение уравнения (1) можно записать следующим образом: где K a известно как константа диссоциации или константа равновесия индикатора, а [In — ] и [HIn] известны как концентрация основных и кислотных форм индикатора, соответственно:
Уравнение можно записать как
Значения отношения были определены из спектрофотометрических измерений, выполненных на двух длинах волн ( λ 1 и λ 2 ) чтобы построить график зависимости pH от.
Согласно закону Бера абсорбция при λ 1 и λ 2 was где абсорбция, молярная абсорбционная способность и длина пути клетки.
При любом pH общая концентрация () как In —, так и HIn была постоянной, и сумма индивидуальных концентраций обеих форм:
В растворе с низким pH все индикаторы находятся в форме. В результате в сильнокислом растворе и
В растворе с высоким pH все индикаторы имеют вид [In — ].В результате в высокоосновном решении
и
Наконец, соотношение может быть определено путем деления отношения уравнений (5) — (8) на соотношение уравнений (6) — (9) [11, 12]:
3. Результаты и обсуждение
Все экстракты цветов имели разную окраску в кислых и основных растворах и имели разные самые высокие пики (макс. Лямбда) в сильнокислой и сильнощелочной среде (таблица 2). Было обнаружено, что конечные точки, определенные для кислотно-основного титрования с использованием экстрактов цветов в качестве индикаторов, очень похожи на таковые для стандартных индикаторов, используемых в лабораториях (таблица 3).
Индикатор
Титрант / титрант
Изменение цвета
Требуемый объем титранта
90OH215 Цвет фторсодержащего водорода 8,87 ± 0,0577
NH 4 OH / HCl
От розового до бесцветного
14,57 ± 0,0577
CH 3 COOH / NaOH
От бесцветного до розового
.07 ± 0,0577
s Метиловый оранжевый
NaOH / HCl
От желтого до красного
9,17 ± 0,0577
NH 4 OH / HCl 905 от желтого до красного
CH 3 COOH / NaOH
От красного до желтого
5,27 ± 0,0577
Метиловый красный
NaOH / HCl
67 От желтого до желтого 6797 ± 0,0577
NH 4 OH / HCl
От желтого к красному
16,13 ± 0,0577
CH 3 COOH / NaOH
От красного к желтому
902,13 ± 0,0577
Bougainvillea glabra
NaOH / HCl
От коричневато-желтого до красного
8,97 ± 0,0577
NH 4 OH / HCl
5 от коричневато-желтого до красного.17 ± 0,0577
CH 3 COOH / NaOH
От красного до коричневато-желтого
12,23 ± 0,0577
Impatiens balsamina
NaOH до красного цвета
9,07 ± 0,0577
NH 4 OH / HCl
От коричневато-желтого до красного
15,3 ± 0,1
CH 3 COOH / NaOH
От красного до коричневато-желтого 12220
.13 ± 0,0577
Bauhinia purpurea
NaOH / HCl
От желтого к красному
8,93 ± 0,0577
NH 16 4 желтый к красному ± 0,0577
CH 3 COOH / NaOH
От красного к желтому
12,3 ± 0,1
P K извлекает значения 9011 и для всех значений цветов метиловый оранжевый (для сравнения) рассчитывались непосредственно из пересечения графиков, как показано на рисунке 1.Значения R 2 этих трех цветочных экстрактов, которые были выше (0,9551, 0,9569 и 0,9649), чем у индикатора метилового красного (0,9260), указывают на лучшую линейную взаимосвязь для трех индикаторов цветов по сравнению с синтетическим индикатором, используемым для сравнение. Рассчитанные значения p K a и диапазон pH для соответствующих индикаторов приведены в таблице 4.
Индикатор
pKa
Диапазон pH
905
Bougainvillea glabra
6.9966 ≈ 7,0
6,0–8,0
Impatiens balsamina
4,3766 ≈ 4,4
3,4–5,4
Bauhinia purpurea
7,0325
3,4003 ≈ 3,4
2,4–4,4
Изменение требуемых объемов титранта при использовании цветочных индикаторов для титрования сильной кислотой и сильным основанием составляет от 0.04–0,1, тогда как вариация синтетических индикаторов находится в диапазоне от 0,1 до 0,3. Для титрования сильной кислотой и слабым основанием изменение требуемого объема титранта составляло от 0,03 до 0,13, в то время как синтетические индикаторы показывают изменение от 0,46 до 1,56. В случае соединения слабая кислота-сильное основание вариации для синтетических индикаторов были очень высокими, так как индикатор метиловый оранжевый не подходит для титрования слабая кислота-сильное основание. Однако показатели метилового красного, Bauhinia purpurea и Impatiens balsamina показывают значение, очень близкое к стандартному показателю — фенолфталеину.Диапазон pH Bougainvillea glabra и Bauhinia purpurea очень близок к диапазону pH фенолфталеина (8,2–10,0). Метиловый апельсин не смог обнаружить острые конечные точки для слабого кислотно-сильного основания, в то время как экстракты цветов Bougainvillea glabra и Bauhinia purpurea обнаруживают конечные точки более точно, чем синтетические индикаторы.
Эти индикаторы натуральных цветов очень важны в современных лабораториях, когда автоматические титраторы не могут титровать некоторые химически активные жидкости.Углеводород может реагировать с пластиковыми материалами, используемыми в цифровых титраторах, поэтому использовать эти современные цифровые измерители нельзя. Методы, предложенные Американским обществом испытаний и материалов, ASTM D974 и ASTM D5984, используют титрование с помощью цветного индикатора для определения основных компонентов в нефтепродуктах и смазочных материалах. Колориметрическое титрование использует визуальные изменения химического соединения, когда его среда меняется с кислой на щелочную. Другими словами, цвет этого индикатора изменится при pH, соответствующем точке перегиба.Метиловый красный используется в качестве индикатора, меняя свой цвет с пурпурного на желтый при pH, соответствующем точке перегиба. В ASTM D974, как и в D4739, в качестве титранта используется соляная кислота; смесь толуола и изопропилового спирта, содержащая небольшое количество воды, используется в качестве системы растворителей, а p -нафтолбензеин используется в качестве цветного индикатора, который имеет оранжевый цвет по кислоте и зелено-коричневый цвет по основанию [15]. Эти синтетические индикаторы могут быть заменены индикаторами из натуральных цветов даже в современных лабораториях.
4. Выводы
Результаты, полученные в результате настоящего исследования, показывают, что аналитический потенциал экстрактов цветков очень многообещающий, что подтверждается их применением в кислотно-щелочной титриметрии. Было обнаружено, что эти экстракты лучше всего подходят для титрования сильной кислотой и сильным основанием по сравнению со слабым кислотно-сильным основанием с резким и четким изменением цвета от красного до коричневато-желтого для экстракта Bougainvillea glabra , от красного до желтого для Bauhinia. purpurea и от красного до коричневато-желтого для экстракта Impatiens balsamina .Все три экстракта цветов дали отчетливое изменение цвета с помощью кислот и оснований, и изменение цвета поддерживалось с помощью различных кислот и оснований. Резкий контраст между их цветами в кислоте и основе сделал пигмент подходящим для использования в качестве кислотно-основных индикаторов.
Доступность и простая процедура экстракции с отличными характеристиками и точными результатами сделали бы эти индикаторы для натуральных цветов подходящими заменителями синтетических индикаторов, используемых во многих лабораториях и исследовательских институтах.Короче говоря, отрасли, исследовательские лаборатории, школы и химические компании, использующие индикаторы для определения кислотности, щелочности, влажности, степени реакций и т. Д., Сочтут предварительные результаты этого исследования ценными для получения эффективных индикаторов из цветы как заменители или возможная замена стандартных индикаторов. Американское общество испытаний и материалов (ASTM) использует колориметрическое титрование с синтетическими индикаторами в качестве стандартного метода для количественной оценки устойчивости смазки к разрушающему воздействию кислотных компонентов.Эти синтетические индикаторы могут быть заменены индикаторами из натуральных цветов в современных лабораториях. Однако недостатком цветочного экстракта является то, что их нужно готовить в свежем виде, поскольку они подвержены грибковому росту через три дня.
Доступность данных
Данные, использованные для подтверждения результатов этого исследования, включены в статью.
Раскрытие информации
Эта работа была проведена в лаборатории кафедры химии факультета естественных наук Восточного университета, Шри-Ланка.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов.
Общие индикаторы кислотной основы | Наука
Обновлено 1 марта 2020 г.
Розанн Козловски
Рецензент: Лана Бандойм, Б.С.
Кислотные и щелочные индикаторы используются в химии для определения pH вещества. Они меняют свой цвет в зависимости от того, добавлены ли они к кислоте, основанию или нейтральному веществу. Большинство индикаторов являются слабыми кислотами и реагируют на изменение концентрации ионов водорода.
Шкала pH и индикаторы кислоты и щелочи
Диапазон pH составляет от 0 до 14, где 7 является нейтральным. PH выше 7 указывает на основание, а значение ниже 7 указывает на кислоту. Шкала pH является логарифмической, и при изменении каждого отдельного числа на шкале происходит десятикратное изменение кислотности или щелочности (основности).
Кислотные и щелочные индикаторы чувствительны к изменению pH или, точнее, к изменению концентрации ионов водорода , H + в растворе.
Большинство кислотных и основных индикаторов представляют собой большие органические молекулы, которые содержат чередующиеся двойные и одинарные углерод-углеродные связи. В основных растворах кислотные и основные индикаторы становятся ионами, теряя ионы водорода из своего химического состава, обычно из группы ОН. Это изменяет структуру молекулы, заставляя индикаторы поглощать свет с разной длиной волны и менять цвет.
Типы индикаторов
Многие типы индикаторов работают со всей шкалой pH.Важно выбрать тот, который соответствует рассматриваемому диапазону pH. Ниже приведены несколько общих индикаторов.
Лакмусовая бумага
Одним из распространенных индикаторов кислот и оснований является лакмусовая бумага, созданная обработкой фильтровальной бумаги красителем, полученным из лишайников. Раствор, нанесенный на лакмусовую бумагу, либо сохранит цвет лакмусовой бумаги, либо изменит его.
Красная лакмусовая кислота — слабая дипротонная кислота; он может отдавать два атома водорода. При pH ниже 4,5 красная лакмусовая бумажка остается красной, но становится синей в основе.При pH выше 8,3 синяя лакмусовая бумажка остается синей, но становится красной в кислоте.
Лакмусовая бумажка не способна определять значение pH, только различает кислоты и основания . Нейтральная лакмусовая бумага пурпурного цвета.
Индикатор фенолфталеина
Индикатор фенолфталеина представляет собой слабую кислоту, имеющую две разные структуры в зависимости от того, находится ли она в щелочном (розовом) или кислотном (бесцветном) растворе. Раствор щелочи поглощает свет в видимом спектре света, и человеческий глаз воспринимает изменение цвета на розовый при уровне pH 8.2, продолжая становиться ярко-пурпурным при pH 10 и выше.
Фенолфталеин обычно используется в качестве индикатора в экспериментах по кислотно-щелочному титрованию в химической лаборатории. Раствор известной концентрации осторожно добавляют к раствору неизвестной концентрации и индикатора фенолфталеина. Когда раствор превращается из бесцветного в розовый (или наоборот), это означает, что титрование , или точка нейтрализации достигнуты, и можно рассчитать неизвестную концентрацию.
Индикатор бромтимолового синего
Бромтимоловый синий, слабая кислота, чаще всего используется в качестве индикатора для относительно нейтральных растворов — слабых кислот и оснований.Диапазон pH составляет от 6 до 7,6. Раствор выглядит желтым до pH 6, в нейтральном растворе он становится зеленым, а в щелочных растворах с pH 7,6 он становится синим.
В лаборатории бромтимоловый синий часто используется в качестве красителя для биологических препаратов , для тестирования фотосинтеза и может использоваться для определения pH в плавательных бассейнах.
Индикатор метилового красного
В качестве индикатора кислоты и основания метиловый красный становится красным в кислых растворах при pH 4,4 и ниже и желтым при pH 6.2 достигается. Между этими конечными значениями цвета, в диапазоне pH от 4,4 до 6,2, цвет оранжевый.
Метиловый красный может использоваться как индикатор кислоты и основания в лаборатории и как азокраситель, самая большая группа синтетических красителей , , обычно используемых для обработки текстильных изделий.
Универсальный индикатор
Универсальный индикатор — это раствор, содержащий смесь индикаторов, часто фенолфталеина, метилового красного и бромтимолового синего. Определение приблизительного pH достигается добавлением в раствор нескольких капель универсального индикатора.
Красный означает сильную кислоту в диапазоне pH от 1 до 4, тогда как слабая кислота имеет оранжевый оттенок. В нейтральном растворе цвет становится желто-зеленым. Пурпурный указывает на сильное основание, pH выше 11, тогда как слабые основания имеют голубоватый оттенок.
Влияние условий в системе на естественные индикаторы
Этот сайт использует файлы cookie для повышения производительности. Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка вашего браузера для приема файлов cookie
Существует множество причин, по которым cookie не может быть установлен правильно.Ниже приведены наиболее частые причины:
В вашем браузере отключены файлы cookie. Вам необходимо сбросить настройки своего браузера, чтобы он принимал файлы cookie, или чтобы спросить вас, хотите ли вы принимать файлы cookie.
Ваш браузер спрашивает вас, хотите ли вы принимать файлы cookie, и вы отказались.
Чтобы принять файлы cookie с этого сайта, нажмите кнопку «Назад» и примите файлы cookie.
Ваш браузер не поддерживает файлы cookie. Если вы подозреваете это, попробуйте другой браузер.
Дата на вашем компьютере в прошлом.Если часы вашего компьютера показывают дату до 1 января 1970 г.,
браузер автоматически забудет файл cookie. Чтобы исправить это, установите правильное время и дату на своем компьютере.
Вы установили приложение, которое отслеживает или блокирует установку файлов cookie.
Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.
Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу.Чтобы предоставить доступ без файлов cookie
потребует, чтобы сайт создавал новый сеанс для каждой посещаемой страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файле cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в cookie; никакая другая информация не фиксируется.
Как правило, в файле cookie может храниться только информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта.Например, сайт
не может определить ваше имя электронной почты, пока вы не введете его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступа к
остальной части вашего компьютера, и только сайт, который создал файл cookie, может его прочитать.
Химия капусты — поиск кислот и оснований
Ключевые концепции Химия Кислоты Базы Свет
Введение Возможно, вы проводили эксперименты с хорошо обозначенными кислотами и основаниями в школе, но задавались ли вы когда-нибудь вопросом, является ли определенный продукт питания или химическое вещество в доме кислотой или основанием? Вы можете узнать это, используя краснокочанную капусту для приготовления индикаторного раствора.
Когда два или более ингредиента полностью растворяются друг в друге, у вас есть решение. Например, при смешивании соли с водой получается прозрачный раствор, даже если соль есть и раствор имеет соленый вкус. При смешивании с водой химическое вещество «жертвует» заряженную частицу (называемую ионом) раствору — в данном случае ион водорода — или «принимает» один из них, определяет, будет ли это раствор кислотным или основным. Индикатор меняет цвет при воздействии такой смеси в зависимости от того, является ли раствор кислотным или основным.
Фон Кислоты — это растворы, которые теряют ионы водорода и обычно имеют кислый вкус. Кислоты, такие как соки цитрусовых и домашний уксус, являются очень распространенными бытовыми растворами. Основания — это растворы, которые вытягивают ионы водорода из раствора на себя, «принимают» их и обычно кажутся скользкими. Основания имеют множество практических применений. Например, «антациды», такие как TUMS, используются для снижения кислотности в желудке. Другие основы делают полезные бытовые чистящие средства.
Чтобы определить, является ли что-то кислотой или щелочью, вы можете использовать химическое вещество, называемое индикатором. Индикатор меняет цвет, когда сталкивается с кислотой или щелочью. Есть много различных типов индикаторов, одни из которых являются жидкостями, а другие сконцентрированы на маленьких полосках лакмусовой бумаги. Индикаторы могут быть извлечены из множества различных источников, включая пигмент многих растений. Например, краснокочанная капуста содержит молекулу индикаторного пигмента, называемого флавином, который представляет собой тип молекулы, называемый антоцианом.Очень кислые растворы сделают антоцианово-красным, тогда как нейтральные растворы сделают его багровым, а щелочные растворы — зеленовато-желтым. Следовательно, цвет, который меняет раствор антоциана, можно использовать для определения pH раствора — показателя того, насколько щелочной или кислый раствор является.
Материалы • Маленькая краснокочанная капуста • Кастрюля с кипятком • Фильтр • Две большие миски или горшки • Терка • Дозатор столовой • Большая ложка (необязательно) • Три или более маленьких белых бумажных стаканчика (также подойдут маленькие белые бумажные стаканы или посуда) • Очки или другие защитные очки • Лимонный или лаймовый сок • Уксус • Чистящее средство на основе отбеливателя • Другие продукты для тестирования, такие как газированная вода, раствор пищевой соды, яичные белки, помидоры, творог (по желанию)
Подготовка • Натереть на терке небольшую красную капусту.Если вы не хотите тереть всю капусту на терке, достаточно натереть половину кочанной капусты. Переложите мелко натертую на терке капусту в большую миску или кастрюлю. • Вскипятите кастрюлю с водой. Будьте осторожны при обращении с кипящей водой. Влейте кипяток в миску с капустной мякотью, пока вода не покроет капусту. • Оставьте капустную смесь настаиваться, периодически помешивая, пока жидкость не станет комнатной температуры. Это должно занять не менее получаса. Жидкость станет красной или пурпурно-красной. • Поместите ситечко на другую большую миску или кастрюлю и вылейте капустную смесь через ситечко, чтобы удалить мякоть. Нажмите на мякоть в сетчатом фильтре, например, большой ложкой, чтобы выжать из мякоти больше жидкости. • Теперь в миске должна быть только жидкость фиолетового или синего цвета. Это будет ваш индикаторный раствор, который вы будете использовать для проверки pH различных жидкостей. • Дети должны носить защитные очки или другие защитные очки, а взрослые должны соблюдать осторожность и соблюдать осторожность при обращении с отбеливателем и уксусом, поскольку они могут вызвать раздражение глаз и кожи.
Процедура • Наполните небольшой белый бумажный стаканчик, стакан или белую посуду одной столовой ложкой раствора индикатора капусты. Какого цвета ваш индикаторный раствор? • Добавляйте в индикаторный раствор капли сока лимона или лайма до тех пор, пока раствор не изменится в цвете. Аккуратно перемешайте раствор и убедитесь, что цвет не изменился. Какого цвета стал раствор? • Цвет раствора будет меняться в зависимости от его pH: красный цвет означает, что pH равен 2; Фиолетовый означает pH 4; Фиолетовый означает pH 6; Синий означает pH 8; Сине-зеленый означает pH 10; Зеленовато-желтый означает pH 12. • Каков pH раствора сока лимона или лайма в зависимости от его цвета? • В другой маленький белый бумажный стаканчик добавьте одну столовую ложку исходного раствора индикатора капусты. Добавляйте капли уксуса, пока не увидите, как раствор меняет цвет. Какого цвета стал раствор уксуса? Каков pH раствора? • В третий маленький белый бумажный стаканчик добавьте одну столовую ложку исходного раствора индикатора капусты. Обращаясь с ним осторожно, добавляйте капли отбеливающего чистящего средства до тех пор, пока раствор не изменит цвет. Какого цвета стал отбеливающий раствор и что это говорит о его pH? • Если вы хотите проверить pH других продуктов, снова добавьте одну столовую ложку исходного раствора индикатора капусты в небольшой белый бумажный стаканчик и добавляйте капли пищи, пока не увидите, как раствор меняет цвет. Если пища не в жидкой форме, раздавите ее или растворите в небольшом количестве воды перед добавлением в индикаторный раствор. Какого цвета стал раствор и что это говорит о его pH? • Extra: Есть и другие овощи и фрукты, которые также можно использовать для создания индикаторов pH: красный лук, кожура яблок, черника, кожица винограда и сливы. Какие различные источники пигмента дают наилучшие показатели? • Extra: Вы можете использовать индикаторное решение для написания секретных сообщений. Просто используйте крепкий лимонный сок, чтобы написать невидимое сообщение на бумаге, и дайте ему высохнуть. Чтобы раскрыть сообщение, нарисуйте индикатор капусты по бумаге кистью.
Наблюдения и результаты Изменился ли цвет индикаторного раствора, когда вы добавили сок лайма или лимона, уксус и отбеливатель? Указывает ли цвет раствора на то, что сок лайма или лимона и уксус были кислыми (имели более низкий pH) и что отбеливатель был щелочным (с более высоким pH)?
Раствор с pH от 5 до 7 является нейтральным, 8 или выше — это основание, а 4 или ниже — кислота.Сок лайма, лимонный сок и уксус являются кислотами, поэтому они должны были сделать индикаторный раствор красным или пурпурным. Отбеливатель — сильная основа, поэтому он должен был придать раствору индикатора зеленовато-желтый цвет.
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу[1].
Предпосылки создания теории химического строения
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова
Термин «химическое строение» впервые ввёл А. М. Бутлеров 19 сентября 1861 года в докладе «О химическом строении веществ» на химической секции Съезда немецких естествоиспытателей и врачей в Шпейере (опубликованном в том же году на немецком[3] и в следующем — на русском языках[4]). В том же докладе он заложил основы классической теории химического строения. Главные положения этой теории следующие:
атомы в молекулах веществ соединены друг с другом согласно их валентности, порядок распределения связей в молекуле называется химическим строением;
изменение этой последовательности приводит к образованию нового вещества с новыми свойствами;
свойства веществ зависят не только от их качественного и количественного состава, но и от «химического строения», то есть от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильно влияют друг на друга атомы, непосредственно связанные между собой;
атомы в молекулах оказывают влияние друг на друга, и это влияние приводит к химическим изменениям поведения атома;
определить состав и строение химического вещества можно по продуктам химических превращений.
Геометрия молекул
В 1864 году Бутлеров первым объясняет явление изомерии, показав что изомеры — это соединения, обладающие одинаковым элементным составом, но различным химическим строением. В 1874 году возникает стереохимия, или трёхмерная структурная химия в форме постулата Вант-Гоффа о тетраэдрической системе валентностей у атома углерода.
В настоящее время принято различать структурную и пространственную изомерию.
Структурную изомерию подразделяют на изомерию скелета, обусловленную различным порядком связи атомов, образующих скелет молекулы, например в Н-бутане и изобутане, и на изомерию положения одинаковых функциональных групп при одинаковом углеродном скелете молекулы, например в орто- , мета- и пара- изомерах ароматических соединений.
Пространственная изомерия обусловлена существованием стереоизомеров, соединений, имеющих одинаковый порядок связей атомов, но различное пространственное расположение. К видам пространственной изомерии относятся: оптическая изомерия, обусловливающая существование энантиомеров — пары стереоизомеров, представляющих собой зеркальные отражения друг от друга, не совмещаемые в пространстве; диастереомерия, обусловливающая существование изомеров, не являющихся энантиомерами; геометрическая изомерия, обусловливающая цис- и транс- изомеров, свойственных соединениям с двойными связями и малыми циклами.
Электронные интерпретации строения молекул
Строение молекулы H2 по Бору
С открытием в 1897 году электрона (Дж. Томсоном, Э. Вихертом) появились электронные интерпретации строения молекул. Американский физикохимик Г. Льюис в 1912 году предложил электронную теорию химической связи, по которой связь между атомами в молекуле осуществляется обобществлённой электронной парой. Электронная теория химической связи Льюиса стала основой классической теории строения в органической химии, базирующейся на представлении о парной связи между атомами, образованной дублетом электронов. Валентный штрих между символами элементов в молекуле был заменён двумя точками, обозначающими связывающую электронную пару.
В 1913 году датский физик Нильс Бор предложил рассматривать электронную пару в форме вращающегося кольца, плоскость которого перпендикулярна оси молекулы и равноудалена от ядер атомов.
Динамическое равновесие молекулярной системы, по Бору, достигается за счёт баланса сил между силами взаимного отталкивания ядер и силами притяжения ядер к плоскости кольца электронов. Боровская интерпретация строения молекулы учитывала кулоновское отталкивание электронов, в кольце они находятся в диаметрально противоположном положении.
Молекулу метана Нильс Бор описывал следующим образом:
Ядро углерода, заключённое в очень маленькое кольцо из двух электронов, расположено в центре (тетраэдра), а ядро водорода по углам. Химические связи представляют собой четыре двухэлектронных кольца, вращающихся вокруг линий, соединяющих центр с углами[5].
Квантовая природа межатомных сил
Схема диполь-дипольного взаимодействия двух атомов водорода по Лондону
В своём докладе, прочитанном в Королевском колледже в Лондоне в ознаменовании столетия опубликования трудов Дж. Максвелла по электромагнитному излучению, К. Коулсон дал анализ происхождения и сущности межатомных сил, приводящих к образованию молекулы[6]. Коулсон, ссылаясь на работу Лондона, на примере двух атомов водорода показывает «каким образом два нейтральных атома или молекулы могут оказывать притяжение друг к другу на значительном расстоянии».
Ядра A и B двух атомов водорода находятся на расстоянии r друг от друга (рис.). Каждый атом несёт по одном электрону (в P и Q соответственно). Совокупность зарядов +e в A и -e в P приблизительно эквивалентна электрическому диполю, имеющему величину e·AP. Подобным же образом совокупность +e в B и -e в Q приблизительно эквивалентна электрическому диполю e·BQ. Эти два диполя взаимодействуют друг с другом. Общая потенциальная энергия двух диполей, m и m’, находящихся на расстоянии r, равна:
На языке волновой механики это выражение рассматривается как возмущение, действующее на оба атома.
Природа межатомных сил имеет электромагнитный характер и называется диполь-дипольным взаимодействием. Существуют диполь-квадрупольные, квадруполь-квадрупольные и другие взаимодействия, при которых энергия изменяется в зависимости от более высоких степеней 1/r[6].
Распределение электронной плотности в химических соединениях
Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода.
Рис. Модель молекулы водорода и её осевая проекция
Электронная плотность — это плотность вероятности обнаружения электрона в данной точке пространства. Электронная плотность нормирована и, соответственно, указывает на вероятное число электронов в данном элементарном объёме.
Вероятностную (статистическую) интерпретацию волновой функции сформулировал М. Борн в 1926 году как только было опубликована волновое уравнение Шрёдингера. В 1954 году М. Борн удостоен Нобелевской премии по физике с формулировкой «За фундаментальное исследование в области квантовой механики, особенно за статистическую интерпретацию волновой функции».
Расчёт электронной плотности проводят с использованием уравнения Шрёдингера, которое решается аналитически точно для систем, содержащих только один электрон[7].
Получаемая радиальная функция распределения вероятности нахождения электрона в атоме водорода обладает максимумом при α0, как показано на рисунке. Этот наиболее вероятный радиус совпадает с боровским радиусом и принят в качестве атомной единицы для линейных размеров 1 а. е. (бор) = 0,529177·10−10м ≈ 0,529 Å. Более размытое облако плотности вероятности, полученное при решении уравнения Шрёдингера для атома водорода, значительно отличается от боровской модели атома и согласуется с принципами неопределённости Гейзенберга. С учётом статистической интерпретации волновой функции М. Борна и принципа неопределённости Гейзенберга длины диполей AP и BQ взаимодействующих атомов в расчётах Ф. Лондона достаточно размыты. Размыто и электронное кольцо в модели молекулы водорода по Н. Бору до состояния тороидального электронного облака с неопределёнными границами.
Наиболее вероятный радиус электронного кольца (тора) молекулы водорода (re) определяется боровским радиусом (α0) и длиной химической связи (d): re2 = α02 — (d/2)2; re = 0,374 Å.
Благодаря пространственной симметрии дипольный момент молекулы водорода равен нулю, что соответствует её низкой химической активности[8]. Пространственная симметрия электронной плотности сохраняется, если соединяющиеся в молекулу атомы имеют одинаковую энергию ионизации. В этом случае связывающее электронное кольцо располагается на равном расстоянии от каждого из ядер. Если же потенциалы ионизации различны электронная плотность смещается в сторону атома с большим первым потенциалом ионизации[9]. Смещение электронной плотности приводит к асимметрии распределения электрических зарядов в молекуле, молекула становится полярной системой с определённым дипольным моментом.
Приближённые квантовохимические методы расчёта
Поскольку точное решение уравнения Шрёдингера для атомно-молекулярных систем, содержащих два и более электрона, невозможно, предложены приближённые методы расчёта электронной плотности. Все они возникли в 1930-х годах, проделали значительный путь развития и долгое время дополняли друг друга. Наиболее важные из них — теория валентных связей, теория молекулярных орбиталей, теория кристаллического поля, теория функционала плотности.
В рамках теории валентных связей разработана концепция резонанса (Л. Полинг) и родственная ей концепция мезомерии (К. Ингольд). Концепция резонанса рассмотрена на примере молекулярного иона водорода. Л. Полинг показал, что стабильность молекулярного иона водорода:
вызвана резонансом электрона, то есть движением его «взад и вперёд» между двумя ядрами с «резонансной частотой», равной по величине энергии резонанса (50 ккал/моль), делённой на константу Планка h. Для молекулярного иона в основном состоянии эта частота равна 7.1014сек−1[10].
Концепция резонанса дополняла постулаты классической теории химического строения и утверждала, что если для данного соединения классическая теория допускает построение нескольких приемлемых структурных формул, то действительному состоянию молекул этого соединения отвечает не какая-либо одна отдельная формула, а некоторое их сочетание (наложение, резонанс структур)[11].
Мезомерия является теорией электронного строения химических соединений, согласно которой истинное распределение электронной плотности в молекуле является промежуточным между распределениями, представленными несколькими классическими формулами[12].
Обычно рассматривают положительный и отрицательный мезомерные эффекты:
Винилхлорид: +М-эффект
Аллил-катион: −М-эффект
Кампания идеологического вмешательства в теорию химического строения
Кампания началась в 1949 году с публикации статьи В. М. Татевского и М. М. Шахпаронова «Об одной махистской теории в химии и её пропагандистах»[13]. В качестве главного объекта нападения была выбрана теория резонанса Л. Полинга. Было объявлено, что «представления о реальной молекуле как о чём-то среднем между двумя (и более) крайними абстрактными структурами являются буржуазными и поэтому направленными против всего самого „святого“». Были указаны и пропагандисты теории Я. К. Сыркин и М. Е. Дяткина — авторы книги «Химическая связь и строение молекул», в которой нашла отражение теория резонанса.
В воздухе очередной раз запахло инквизицией. В этой тревожной обстановке ведущие химики страны собрались на Всесоюзное совещание по проблемам химического строения (1951 г., Москва). Стенограмма этого совещания — один из наиболее позорных документов, когда-либо созданных коллективными усилиями ученых, хранится в химических библиотеках всего мира, и от этого срама бог весть когда ещё удастся отмыться… До крови не дошло — спасла оттепель, начавшаяся весной следующего года. Я. К. Сыркин и М. Е. Дяткина, подготовленные недавними друзьями и коллегами к выдаче в качестве первых козлов отпущения, уцелели; более того Я. К. Сыркин в дальнейшем стал академиком[14].
Лайнус Полинг удостоен в 1954 году Нобелевской премии по химии «за исследование природы химической связи и её применение для определения структуры сложных соединений».
Однако консенсус в теории химического строения не был достигнут. В. М. Татевский в курсе «Строение молекул» (1977 г.) отмечал:
… полностью выпадают и «висят в воздухе» представления уходящих в прошлое, но всё ещё фигурирующих в литературе так называемых «теории резонанса» и «теории мезомерии», которые не имеют основы ни в классической теории химического строения, ни в законных приложениях классической физики к вопросам строения молекул, ни в квантовой механике[15].
Лишь в 1991 году проведён принципиальный анализ кампании по борьбе с теорией резонанса и было показано, что эта кампания «нанесла серьёзный ущерб престижу советской науки»[16].
Атомы в молекулах
В классической теории химического строения понятие атома в молекуле является изначальным. Интуитивно ясно, что атом в молекуле меняется, меняются и его свойства в зависимости от окружения этого атома, прежде всего ближайшего. Основным показателем является расстояние между атомами в молекуле, определяющее как длину химической связи, так и прочность химической связи.
В квантовой теории понятие атома вторично. Так, по утверждению В. М. Татевского, молекула не состоит из атомов: «С современной точки зрения ясно, что при образовании молекулы сохраняются не атомы, а только ядра атомов и электроны»[17].
Было предпринято множество попыток сохранить понятие атома в молекуле, но, тем не менее, практически всегда они не удовлетворяли последующих исследователей по тем или иным причинам.
Одна из наиболее удачных попыток сохранения классической концепции атома в молекуле принадлежит Р. Бейдеру и его сотрудникам[18]. В рамках этой теории (QTAIM) электронная плотность «задаёт некоторое скалярное поле в трёхмерном пространстве, которое может быть охарактеризовано, например, его совокупностью экстремальных точек, линий и поверхностей, особых точек и т. п.»[19].
Таким образом, в квантовой теории атомов в молекулах Р. Бейдера оказывается возможным физическое обоснование ключевых понятий химии «атом», «молекула», «химическая связь» в терминах топологии функции электронной плотности в трёхмерном пространстве и описание химического строения молекул.
Электронная корреляция и конфигурация молекул
Электронная корреляция (взаимная обусловленность движения всех электронов атомной или молекулярной системы как целого определяется электростатическим отталкиванием электронов и статическим особенностями систем, в частности принципом Паули (фермиевская корреляция). Полный учёт электронной корреляции при расчёте энергии и определении электронной структуры системы достигается методом конфигурационного взаимодействия.
Простая и надёжная система правил для понимания и предсказания конфигурации молекул заложена в теории отталкивания электронных пар, наиболее важное правило которой достаточно эмпирично, хотя имеет квантовомеханическое обоснование, заключающееся в принципе Паули, а именно «электронные пары принимают такое расположение на валентной оболочке атома, при котором они максимально удалены друг от друга». При этом конфигурация молекулы будет определяться числом связывающих и неподелённых электронных пар у центрального атома:
две связывающие электронные пары дают линейную конфигурацию молекулы;
три — конфигурацию правильного треугольника;
четыре — конфигурацию тетраэдра;
пять — тригонально-бипирамидальную конфигурацию;
шесть — конфигурацию октаэдра.
Наличие неподелённых электронных пар у центрального атома приводит к расширению типов конфигураций молекулы[20].
Открытие Н. Бартлеттом в 1962 году первого соединения ксенона положило начало интенсивному развитию химии благородных газов, что привело к получению большого ряда соединений инертных газов и установлению их химического строения.
Оказалось, что все соединения благородных газов имеют обыкновенные ковалентные связи и общепринятые конфигурации молекул. Так, конфигурация молекулы дифторида ксенона линейна: связи ксенон—фтор являются аксиальными и взаимодействуют с тремя неподелёнными электронными парами, находящимися в экваториальных положениях.
В молекуле тетрафторида ксенона осуществляется конфигурация с плоским квадратным расположением лигандов. Неподелённые электронные пары ксенона максимально удалены друг от друга и расположены по разные стороны от этого квадрата.
Структура молекулы гексафторида ксенона представляет собой искажённый октаэдр. «Отклонение атомов фтора от вершин правильного октаэдра указывает на то, что неподелённая электронная пара занимает положение над центром одной из граней октаэдра, а атомы фтора, расположенные в углах этой грани, вынуждены раздвинуться»[20].
Открытие Бартлетта показало ошибочность популярного в то время правила октета,[как?] согласно которому при образовании молекул атомы удовлетворяют свою потребность в достижении 8-электронной валентной оболочки, подобной электронной конфигурации благородных газов за счёт попарного обобществления своих валентных электронов. Оказалось, что атомы благородных газов, имея полностью заполненную валентную оболочку, могут вступать в химические реакции и участвовать в химическом строении молекул.
Строение электронодефицитных соединений
Структурная проблема, связанная с электронодефицитными соединениями, довольно сложна. Фундаментальная трудность заключалась в том, что в молекулах нет достаточного числа валентных электронов для того, чтобы связать все атомы обычными двухэлектронными связями. Так, например, в молекуле диборана имеется двенадцать валентных электронов, все двенадцать нужны для образования ковалентных связей шести атомов водорода с бором, так что для связи атомов бора между собой электронов не остаётся. Сам Полинг допускал, что в диборане функционируют одноэлектронные связи, а молекула в основном состоянии резонирует между семью структурами льюисовского типа, а также между многочисленными структурами, содержащими одноэлектронные связи.
Однако заслуженное признание получили исследования природы химической связи в бороводородах, выполненные американским физикохимиком У. Липскомбом. В его интерпретации в диборане имеют место четыре двух- и две трёхцентровые связи.
Четыре концевые двухцентровые двухэлектронные связи HB лежат в одной плоскости. Два же центральных атома водорода расположены симметрично над этой плоскостью и под нею и объединены с атомами бора двумя трёхцентровыми связями.
В 1976 году Липскомб удостоен Нобелевской премии по химии с формулировкой «за исследование структуры боранов (боргидритов), проясняющих проблему химических связей».
Особенности строения сэндвичевых соединений
Молекула ферроцена
Дальнейшее развитие теории химического строения связано с открытием и установлением структуры ферроцена[21]. Оказалось, что при взаимодействии хлорида двухвалентного железа и циклопентадиена вместо ожидаемого соединения с двумя химическими связями железо-углерод образуется сэндвичеобразное соединение, в котором все 10 атомов углерода соединены с атомом железа.
Рентгеноструктурными исследованиями Э. Фишера установлено, что все атомы углерода в молекуле ферроцена структурно и химически эквивалентны. Атом металла взаимодействует не с одним или двумя конкретными атомами углерода, а со всеми атомами углерода двух циклопентадиенильных колец сразу. Атом металла как бы завис в пространстве между двумя циклами, представленных параллельными правильными пятиугольниками.
В настоящее время получены металлоцены — бициклопентадиенильные соединения переходных металлов для большинства d-элементов.
Сэндвичевые соединения образуются также с использованием в качестве органического фрагмента бензола или циклооктатетраена. Так, например, в ураноцене (см. рисунок) атом урана удерживает два восьмичленных кольца. Все 16 связей уран—углерод в ураноцене идентичны.
В 1973 году Э. Фишер и Дж. Уилкинсон удостоены Нобелевской премии по химии с формулировкой «За новаторскую, проделанную независимо друг от друга, работу в области металлорганических, так называемых сэндвичевых соединений».
Считается, что в сэндвичевых соединениях реализуется многоцентровая связь, тип химической связи, в которой в отличие от двухцентровой двухэлектронной связи, связывающие электронные пары распределены в пространстве трех или большего числа атомных центров молекулы, иона, радикала.[22]
Примечания
↑ 12 Совещание по теории химического строения в органической химии. Успехи физических наук.. — 1951, октябрь. — Т. XLV, вып. 2. — С. 277—293.
↑ Химический энциклопедический словарь / гл. ред. И. Л. Кнунянц. — Сов. энциклопедия, 1983. — С. 652. — 792 с.
↑ Butlerow A. (1861). «Einiges über die chemische Structur der Körper. (Vorgetragen in der chemischen Section der 36. Versammlung deutscher Naturforscher und Aerzte zu Speyer am 19.Septbr.)». Zeitschrift für Chemie und Pharmacie4: 549-560.
↑ Бутлеров А. О химическом строении веществ // Учёные записки Казанского университета (отд. физ.-мат. и мед. наук). Вып.1, отд.1. — 1862. — С. 1—11.
↑ Бор Н. Избранные научные труды. — М.: «Наука», 1970. — Т. 1. — С. 147. — 584 с.
↑ 123Коулсон К. Успехи физических наук (статья «Межатомные силы — от Максвелла до Шрёдингера»). — 1963, ноябрь. — Т. LXXXI, вып.3. — С. 545-556.
↑ Шусторович Е. М. Химическая связь. Сущность и проблемы.. — М.: «Наука», 1973. — С. 6. — 232 с.
↑ Потапов А. А. Ренессанс классического атома. — М.: «Наука», 2011. — С. 352. — 444 с.
↑ Ганкин В. Ю., Ганкин Ю. В. Как образуется химическая связь и протекают химические реакции. — М.: «Граница». — С. 65. — 320 с.
↑ Паулинг Л. Природа химической связи. — М.-Л.: Химическая литература, 1947. — С. 25—26. — 440 с.
↑ Новая иллюстрированная энциклопедия. Пр-Ро. Кн.15. — М.: Большая российская энциклопедия, 2002. — С. 169. — 255 с.
↑ Новая иллюстрированная энциклопедия. Ма-Мо. Кн.11. — М.: Большая российская энциклопедия, 2002. — С. 141. — 255 с.
↑ Вопросы философии. — 1949. — № 3. — С. 176—192.
↑ Охлобыстин О. Ю. Жизнь и смерть химических идей. Очерки по истории теоретической химии.. — М.: Наука, 1989. — С. 184. — 190 с.
↑ Татевский В. М. Строение молекул.. — М.: Химия, 1977. — С. 10. — 512 с.
↑ Сонин А. С. Печальный юбилей одной кампании. (рус.) // Вестник РАН. — 1991. — Т. 61, № 8. — С. 96—107. Архивировано 5 мая 2010 года.
↑ Татевский В. М. Квантовая механика и теория строения молекул. — М.: Издательство МГУ, 1965. — С. 17. — 162 с.
↑ Бейдер Р. Атомы в молекулах. Квантовая теория. — М.: Мир, 2001. — 532 с.
↑ Степанов Н. Ф. Квантовая механика и квантовая химия. — М.: Мир, 2001. — С. 487. — 519 с.
↑ 12Р. Гиллеспи. Геометрия молекул. — М.: Мир, 1975. — 278 с.
↑ Несмеянов А.Н. Ферроцен и родственные соединения. Избранные труды 1969 — 1979. — М.: Советская энциклопедия, 1982.
↑
Химический Энциклопедический словарь. — М.: Советская энциклопедия, 1983. — 792 с.
wikipedia.green
Теория химического строения — Википедия. Что такое Теория химического строения
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу[1].
Предпосылки создания теории химического строения
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова
Термин «химическое строение» впервые ввёл А. М. Бутлеров 19 сентября 1861 года в докладе «О химическом строении веществ» на химической секции Съезда немецких естествоиспытателей и врачей в Шпейере (опубликованном в том же году на немецком[3] и в следующем — на русском языках[4]). В том же докладе он заложил основы классической теории химического строения. Главные положения этой теории следующие:
атомы в молекулах веществ соединены друг с другом согласно их валентности, порядок распределения связей в молекуле называется химическим строением;
изменение этой последовательности приводит к образованию нового вещества с новыми свойствами;
свойства веществ зависят не только от их качественного и количественного состава, но и от «химического строения», то есть от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильно влияют друг на друга атомы, непосредственно связанные между собой;
атомы в молекулах оказывают влияние друг на друга, и это влияние приводит к химическим изменениям поведения атома;
определить состав и строение химического вещества можно по продуктам химических превращений.
Геометрия молекул
В 1864 году Бутлеров первым объясняет явление изомерии, показав что изомеры — это соединения, обладающие одинаковым элементным составом, но различным химическим строением. В 1874 году возникает стереохимия, или трёхмерная структурная химия в форме постулата Вант-Гоффа о тетраэдрической системе валентностей у атома углерода.
В настоящее время принято различать структурную и пространственную изомерию.
Структурную изомерию подразделяют на изомерию скелета, обусловленную различным порядком связи атомов, образующих скелет молекулы, например в Н-бутане и изобутане, и на изомерию положения одинаковых функциональных групп при одинаковом углеродном скелете молекулы, например в орто- , мета- и пара- изомерах ароматических соединений.
Пространственная изомерия обусловлена существованием стереоизомеров, соединений, имеющих одинаковый порядок связей атомов, но различное пространственное расположение. К видам пространственной изомерии относятся: оптическая изомерия, обусловливающая существование энантиомеров — пары стереоизомеров, представляющих собой зеркальные отражения друг от друга, не совмещаемые в пространстве; диастереомерия, обусловливающая существование изомеров, не являющихся энантиомерами; геометрическая изомерия, обусловливающая цис- и транс- изомеров, свойственных соединениям с двойными связями и малыми циклами.
Электронные интерпретации строения молекул
Строение молекулы H2 по Бору
С открытием в 1897 году электрона (Дж. Томсоном, Э. Вихертом) появились электронные интерпретации строения молекул. Американский физикохимик Г. Льюис в 1912 году предложил электронную теорию химической связи, по которой связь между атомами в молекуле осуществляется обобществлённой электронной парой. Электронная теория химической связи Льюиса стала основой классической теории строения в органической химии, базирующейся на представлении о парной связи между атомами, образованной дублетом электронов. Валентный штрих между символами элементов в молекуле был заменён двумя точками, обозначающими связывающую электронную пару.
В 1913 году датский физик Нильс Бор предложил рассматривать электронную пару в форме вращающегося кольца, плоскость которого перпендикулярна оси молекулы и равноудалена от ядер атомов.
Динамическое равновесие молекулярной системы, по Бору, достигается за счёт баланса сил между силами взаимного отталкивания ядер и силами притяжения ядер к плоскости кольца электронов. Боровская интерпретация строения молекулы учитывала кулоновское отталкивание электронов, в кольце они находятся в диаметрально противоположном положении.
Молекулу метана Нильс Бор описывал следующим образом:
Ядро углерода, заключённое в очень маленькое кольцо из двух электронов, расположено в центре (тетраэдра), а ядро водорода по углам. Химические связи представляют собой четыре двухэлектронных кольца, вращающихся вокруг линий, соединяющих центр с углами[5].
Квантовая природа межатомных сил
Схема диполь-дипольного взаимодействия двух атомов водорода по Лондону
В своём докладе, прочитанном в Королевском колледже в Лондоне в ознаменовании столетия опубликования трудов Дж. Максвелла по электромагнитному излучению, К. Коулсон дал анализ происхождения и сущности межатомных сил, приводящих к образованию молекулы[6]. Коулсон, ссылаясь на работу Лондона, на примере двух атомов водорода показывает «каким образом два нейтральных атома или молекулы могут оказывать притяжение друг к другу на значительном расстоянии».
Ядра A и B двух атомов водорода находятся на расстоянии r друг от друга (рис.). Каждый атом несёт по одном электрону (в P и Q соответственно). Совокупность зарядов +e в A и -e в P приблизительно эквивалентна электрическому диполю, имеющему величину e·AP. Подобным же образом совокупность +e в B и -e в Q приблизительно эквивалентна электрическому диполю e·BQ. Эти два диполя взаимодействуют друг с другом. Общая потенциальная энергия двух диполей, m и m’, находящихся на расстоянии r, равна:
На языке волновой механики это выражение рассматривается как возмущение, действующее на оба атома.
Природа межатомных сил имеет электромагнитный характер и называется диполь-дипольным взаимодействием. Существуют диполь-квадрупольные, квадруполь-квадрупольные и другие взаимодействия, при которых энергия изменяется в зависимости от более высоких степеней 1/r[6].
Распределение электронной плотности в химических соединениях
Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода.
Рис. Модель молекулы водорода и её осевая проекция
Электронная плотность — это плотность вероятности обнаружения электрона в данной точке пространства. Электронная плотность нормирована и, соответственно, указывает на вероятное число электронов в данном элементарном объёме.
Вероятностную (статистическую) интерпретацию волновой функции сформулировал М. Борн в 1926 году как только было опубликована волновое уравнение Шрёдингера. В 1954 году М. Борн удостоен Нобелевской премии по физике с формулировкой «За фундаментальное исследование в области квантовой механики, особенно за статистическую интерпретацию волновой функции».
Расчёт электронной плотности проводят с использованием уравнения Шрёдингера, которое решается аналитически точно для систем, содержащих только один электрон[7].
Получаемая радиальная функция распределения вероятности нахождения электрона в атоме водорода обладает максимумом при α0, как показано на рисунке. Этот наиболее вероятный радиус совпадает с боровским радиусом и принят в качестве атомной единицы для линейных размеров 1 а. е. (бор) = 0,529177·10−10м ≈ 0,529 Å. Более размытое облако плотности вероятности, полученное при решении уравнения Шрёдингера для атома водорода, значительно отличается от боровской модели атома и согласуется с принципами неопределённости Гейзенберга. С учётом статистической интерпретации волновой функции М. Борна и принципа неопределённости Гейзенберга длины диполей AP и BQ взаимодействующих атомов в расчётах Ф. Лондона достаточно размыты. Размыто и электронное кольцо в модели молекулы водорода по Н. Бору до состояния тороидального электронного облака с неопределёнными границами.
Наиболее вероятный радиус электронного кольца (тора) молекулы водорода (re) определяется боровским радиусом (α0) и длиной химической связи (d): re2 = α02 — (d/2)2; re = 0,374 Å.
Благодаря пространственной симметрии дипольный момент молекулы водорода равен нулю, что соответствует её низкой химической активности[8]. Пространственная симметрия электронной плотности сохраняется, если соединяющиеся в молекулу атомы имеют одинаковую энергию ионизации. В этом случае связывающее электронное кольцо располагается на равном расстоянии от каждого из ядер. Если же потенциалы ионизации различны электронная плотность смещается в сторону атома с большим первым потенциалом ионизации[9]. Смещение электронной плотности приводит к асимметрии распределения электрических зарядов в молекуле, молекула становится полярной системой с определённым дипольным моментом.
Приближённые квантовохимические методы расчёта
Поскольку точное решение уравнения Шрёдингера для атомно-молекулярных систем, содержащих два и более электрона, невозможно, предложены приближённые методы расчёта электронной плотности. Все они возникли в 1930-х годах, проделали значительный путь развития и долгое время дополняли друг друга. Наиболее важные из них — теория валентных связей, теория молекулярных орбиталей, теория кристаллического поля, теория функционала плотности.
В рамках теории валентных связей разработана концепция резонанса (Л. Полинг) и родственная ей концепция мезомерии (К. Ингольд). Концепция резонанса рассмотрена на примере молекулярного иона водорода. Л. Полинг показал, что стабильность молекулярного иона водорода:
вызвана резонансом электрона, то есть движением его «взад и вперёд» между двумя ядрами с «резонансной частотой», равной по величине энергии резонанса (50 ккал/моль), делённой на константу Планка h. Для молекулярного иона в основном состоянии эта частота равна 7.1014сек−1[10].
Концепция резонанса дополняла постулаты классической теории химического строения и утверждала, что если для данного соединения классическая теория допускает построение нескольких приемлемых структурных формул, то действительному состоянию молекул этого соединения отвечает не какая-либо одна отдельная формула, а некоторое их сочетание (наложение, резонанс структур)[11].
Мезомерия является теорией электронного строения химических соединений, согласно которой истинное распределение электронной плотности в молекуле является промежуточным между распределениями, представленными несколькими классическими формулами[12].
Обычно рассматривают положительный и отрицательный мезомерные эффекты:
Винилхлорид: +М-эффект
Аллил-катион: −М-эффект
Кампания идеологического вмешательства в теорию химического строения
Кампания началась в 1949 году с публикации статьи В. М. Татевского и М. М. Шахпаронова «Об одной махистской теории в химии и её пропагандистах»[13]. В качестве главного объекта нападения была выбрана теория резонанса Л. Полинга. Было объявлено, что «представления о реальной молекуле как о чём-то среднем между двумя (и более) крайними абстрактными структурами являются буржуазными и поэтому направленными против всего самого „святого“». Были указаны и пропагандисты теории Я. К. Сыркин и М. Е. Дяткина — авторы книги «Химическая связь и строение молекул», в которой нашла отражение теория резонанса.
В воздухе очередной раз запахло инквизицией. В этой тревожной обстановке ведущие химики страны собрались на Всесоюзное совещание по проблемам химического строения (1951 г., Москва). Стенограмма этого совещания — один из наиболее позорных документов, когда-либо созданных коллективными усилиями ученых, хранится в химических библиотеках всего мира, и от этого срама бог весть когда ещё удастся отмыться… До крови не дошло — спасла оттепель, начавшаяся весной следующего года. Я. К. Сыркин и М. Е. Дяткина, подготовленные недавними друзьями и коллегами к выдаче в качестве первых козлов отпущения, уцелели; более того Я. К. Сыркин в дальнейшем стал академиком[14].
Лайнус Полинг удостоен в 1954 году Нобелевской премии по химии «за исследование природы химической связи и её применение для определения структуры сложных соединений».
Однако консенсус в теории химического строения не был достигнут. В. М. Татевский в курсе «Строение молекул» (1977 г.) отмечал:
… полностью выпадают и «висят в воздухе» представления уходящих в прошлое, но всё ещё фигурирующих в литературе так называемых «теории резонанса» и «теории мезомерии», которые не имеют основы ни в классической теории химического строения, ни в законных приложениях классической физики к вопросам строения молекул, ни в квантовой механике[15].
Лишь в 1991 году проведён принципиальный анализ кампании по борьбе с теорией резонанса и было показано, что эта кампания «нанесла серьёзный ущерб престижу советской науки»[16].
Атомы в молекулах
В классической теории химического строения понятие атома в молекуле является изначальным. Интуитивно ясно, что атом в молекуле меняется, меняются и его свойства в зависимости от окружения этого атома, прежде всего ближайшего. Основным показателем является расстояние между атомами в молекуле, определяющее как длину химической связи, так и прочность химической связи.
В квантовой теории понятие атома вторично. Так, по утверждению В. М. Татевского, молекула не состоит из атомов: «С современной точки зрения ясно, что при образовании молекулы сохраняются не атомы, а только ядра атомов и электроны»[17].
Было предпринято множество попыток сохранить понятие атома в молекуле, но, тем не менее, практически всегда они не удовлетворяли последующих исследователей по тем или иным причинам.
Одна из наиболее удачных попыток сохранения классической концепции атома в молекуле принадлежит Р. Бейдеру и его сотрудникам[18]. В рамках этой теории (QTAIM) электронная плотность «задаёт некоторое скалярное поле в трёхмерном пространстве, которое может быть охарактеризовано, например, его совокупностью экстремальных точек, линий и поверхностей, особых точек и т. п.»[19].
Таким образом, в квантовой теории атомов в молекулах Р. Бейдера оказывается возможным физическое обоснование ключевых понятий химии «атом», «молекула», «химическая связь» в терминах топологии функции электронной плотности в трёхмерном пространстве и описание химического строения молекул.
Электронная корреляция и конфигурация молекул
Электронная корреляция (взаимная обусловленность движения всех электронов атомной или молекулярной системы как целого определяется электростатическим отталкиванием электронов и статическим особенностями систем, в частности принципом Паули (фермиевская корреляция). Полный учёт электронной корреляции при расчёте энергии и определении электронной структуры системы достигается методом конфигурационного взаимодействия.
Простая и надёжная система правил для понимания и предсказания конфигурации молекул заложена в теории отталкивания электронных пар, наиболее важное правило которой достаточно эмпирично, хотя имеет квантовомеханическое обоснование, заключающееся в принципе Паули, а именно «электронные пары принимают такое расположение на валентной оболочке атома, при котором они максимально удалены друг от друга». При этом конфигурация молекулы будет определяться числом связывающих и неподелённых электронных пар у центрального атома:
две связывающие электронные пары дают линейную конфигурацию молекулы;
три — конфигурацию правильного треугольника;
четыре — конфигурацию тетраэдра;
пять — тригонально-бипирамидальную конфигурацию;
шесть — конфигурацию октаэдра.
Наличие неподелённых электронных пар у центрального атома приводит к расширению типов конфигураций молекулы[20].
Открытие Н. Бартлеттом в 1962 году первого соединения ксенона положило начало интенсивному развитию химии благородных газов, что привело к получению большого ряда соединений инертных газов и установлению их химического строения.
Оказалось, что все соединения благородных газов имеют обыкновенные ковалентные связи и общепринятые конфигурации молекул. Так, конфигурация молекулы дифторида ксенона линейна: связи ксенон—фтор являются аксиальными и взаимодействуют с тремя неподелёнными электронными парами, находящимися в экваториальных положениях.
В молекуле тетрафторида ксенона осуществляется конфигурация с плоским квадратным расположением лигандов. Неподелённые электронные пары ксенона максимально удалены друг от друга и расположены по разные стороны от этого квадрата.
Структура молекулы гексафторида ксенона представляет собой искажённый октаэдр. «Отклонение атомов фтора от вершин правильного октаэдра указывает на то, что неподелённая электронная пара занимает положение над центром одной из граней октаэдра, а атомы фтора, расположенные в углах этой грани, вынуждены раздвинуться»[20].
Открытие Бартлетта показало ошибочность популярного в то время правила октета,[как?] согласно которому при образовании молекул атомы удовлетворяют свою потребность в достижении 8-электронной валентной оболочки, подобной электронной конфигурации благородных газов за счёт попарного обобществления своих валентных электронов. Оказалось, что атомы благородных газов, имея полностью заполненную валентную оболочку, могут вступать в химические реакции и участвовать в химическом строении молекул.
Строение электронодефицитных соединений
Структурная проблема, связанная с электронодефицитными соединениями, довольно сложна. Фундаментальная трудность заключалась в том, что в молекулах нет достаточного числа валентных электронов для того, чтобы связать все атомы обычными двухэлектронными связями. Так, например, в молекуле диборана имеется двенадцать валентных электронов, все двенадцать нужны для образования ковалентных связей шести атомов водорода с бором, так что для связи атомов бора между собой электронов не остаётся. Сам Полинг допускал, что в диборане функционируют одноэлектронные связи, а молекула в основном состоянии резонирует между семью структурами льюисовского типа, а также между многочисленными структурами, содержащими одноэлектронные связи.
Однако заслуженное признание получили исследования природы химической связи в бороводородах, выполненные американским физикохимиком У. Липскомбом. В его интерпретации в диборане имеют место четыре двух- и две трёхцентровые связи.
Четыре концевые двухцентровые двухэлектронные связи HB лежат в одной плоскости. Два же центральных атома водорода расположены симметрично над этой плоскостью и под нею и объединены с атомами бора двумя трёхцентровыми связями.
В 1976 году Липскомб удостоен Нобелевской премии по химии с формулировкой «за исследование структуры боранов (боргидритов), проясняющих проблему химических связей».
Особенности строения сэндвичевых соединений
Молекула ферроцена
Дальнейшее развитие теории химического строения связано с открытием и установлением структуры ферроцена[21]. Оказалось, что при взаимодействии хлорида двухвалентного железа и циклопентадиена вместо ожидаемого соединения с двумя химическими связями железо-углерод образуется сэндвичеобразное соединение, в котором все 10 атомов углерода соединены с атомом железа.
Рентгеноструктурными исследованиями Э. Фишера установлено, что все атомы углерода в молекуле ферроцена структурно и химически эквивалентны. Атом металла взаимодействует не с одним или двумя конкретными атомами углерода, а со всеми атомами углерода двух циклопентадиенильных колец сразу. Атом металла как бы завис в пространстве между двумя циклами, представленных параллельными правильными пятиугольниками.
В настоящее время получены металлоцены — бициклопентадиенильные соединения переходных металлов для большинства d-элементов.
Сэндвичевые соединения образуются также с использованием в качестве органического фрагмента бензола или циклооктатетраена. Так, например, в ураноцене (см. рисунок) атом урана удерживает два восьмичленных кольца. Все 16 связей уран—углерод в ураноцене идентичны.
В 1973 году Э. Фишер и Дж. Уилкинсон удостоены Нобелевской премии по химии с формулировкой «За новаторскую, проделанную независимо друг от друга, работу в области металлорганических, так называемых сэндвичевых соединений».
Считается, что в сэндвичевых соединениях реализуется многоцентровая связь, тип химической связи, в которой в отличие от двухцентровой двухэлектронной связи, связывающие электронные пары распределены в пространстве трех или большего числа атомных центров молекулы, иона, радикала.[22]
Примечания
↑ 12 Совещание по теории химического строения в органической химии. Успехи физических наук.. — 1951, октябрь. — Т. XLV, вып. 2. — С. 277—293.
↑ Химический энциклопедический словарь / гл. ред. И. Л. Кнунянц. — Сов. энциклопедия, 1983. — С. 652. — 792 с.
↑ Butlerow A. (1861). «Einiges über die chemische Structur der Körper. (Vorgetragen in der chemischen Section der 36. Versammlung deutscher Naturforscher und Aerzte zu Speyer am 19.Septbr.)». Zeitschrift für Chemie und Pharmacie4: 549-560.
↑ Бутлеров А. О химическом строении веществ // Учёные записки Казанского университета (отд. физ.-мат. и мед. наук). Вып.1, отд.1. — 1862. — С. 1—11.
↑ Бор Н. Избранные научные труды. — М.: «Наука», 1970. — Т. 1. — С. 147. — 584 с.
↑ 123Коулсон К. Успехи физических наук (статья «Межатомные силы — от Максвелла до Шрёдингера»). — 1963, ноябрь. — Т. LXXXI, вып.3. — С. 545-556.
↑ Шусторович Е. М. Химическая связь. Сущность и проблемы.. — М.: «Наука», 1973. — С. 6. — 232 с.
↑ Потапов А. А. Ренессанс классического атома. — М.: «Наука», 2011. — С. 352. — 444 с.
↑ Ганкин В. Ю., Ганкин Ю. В. Как образуется химическая связь и протекают химические реакции. — М.: «Граница». — С. 65. — 320 с.
↑ Паулинг Л. Природа химической связи. — М.-Л.: Химическая литература, 1947. — С. 25—26. — 440 с.
↑ Новая иллюстрированная энциклопедия. Пр-Ро. Кн.15. — М.: Большая российская энциклопедия, 2002. — С. 169. — 255 с.
↑ Новая иллюстрированная энциклопедия. Ма-Мо. Кн.11. — М.: Большая российская энциклопедия, 2002. — С. 141. — 255 с.
↑ Вопросы философии. — 1949. — № 3. — С. 176—192.
↑ Охлобыстин О. Ю. Жизнь и смерть химических идей. Очерки по истории теоретической химии.. — М.: Наука, 1989. — С. 184. — 190 с.
↑ Татевский В. М. Строение молекул.. — М.: Химия, 1977. — С. 10. — 512 с.
↑ Сонин А. С. Печальный юбилей одной кампании. (рус.) // Вестник РАН. — 1991. — Т. 61, № 8. — С. 96—107. Архивировано 5 мая 2010 года.
↑ Татевский В. М. Квантовая механика и теория строения молекул. — М.: Издательство МГУ, 1965. — С. 17. — 162 с.
↑ Бейдер Р. Атомы в молекулах. Квантовая теория. — М.: Мир, 2001. — 532 с.
↑ Степанов Н. Ф. Квантовая механика и квантовая химия. — М.: Мир, 2001. — С. 487. — 519 с.
↑ 12Р. Гиллеспи. Геометрия молекул. — М.: Мир, 1975. — 278 с.
↑ Несмеянов А.Н. Ферроцен и родственные соединения. Избранные труды 1969 — 1979. — М.: Советская энциклопедия, 1982.
↑
Химический Энциклопедический словарь. — М.: Советская энциклопедия, 1983. — 792 с.
wiki.sc
Теория химического строения Википедия
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу[1].
Предпосылки создания теории химического строения[ | ]
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова[ |
ru-wiki.ru
Теория химического строения Википедия
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу[1].
Предпосылки создания теории химического строения
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова
Термин «химическое строение» впервые ввёл А. М. Бутлеров 19 сентября 1861 года в докладе «О химическом строении веществ» на химической секции Съезда немецких естествоиспытателей и врачей в Шпейере (опубликованном в том же году на немецком[3] и в следующем — на русском языках[4]). В том же докладе он заложил основы классической теории химического строения. Главные положения этой теории следующие:
атомы в молекулах веществ соединены друг с другом согласно их валентности, порядок распределения связей в молекуле называется химическим строением;
изменение этой последовательности приводит к образованию нового вещества с новыми свойствами;
свойства веществ зависят не только от их качественного и количественного состава, но и от «химического строения», то есть от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильно влияют друг на друга атомы, непосредственно связанные между собой;
атомы в молекулах оказывают влияние друг на друга, и это влияние приводит к химическим изменениям поведения атома;
определить состав и строение химического вещества можно по продуктам химических превращений.
Геометрия молекул
В 1864 году Бутлеров первым объясняет явление изомерии, показав что изомеры — это соединения, обладающие одинаковым элементным составом, но различным химическим строением. В 1874 году возникает стереохимия, или трёхмерная структурная химия в форме постулата Вант-Гоффа о тетраэдрической системе валентностей у атома углерода.
В настоящее время принято различать структурную и пространственную изомерию.
Структурную изомерию подразделяют на изомерию скелета, обусловленную различным порядком связи атомов, образующих скелет молекулы, например в Н-бутане и изобутане, и на изомерию положения одинаковых функциональных групп при одинаковом углеродном скелете молекулы, например в орто- , мета- и пара- изомерах ароматических соединений.
Пространственная изомерия обусловлена существование
ruwikiorg.ru
Теория химического строения — WiKi
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу[1].
Предпосылки создания теории химического строения
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова
Термин «химическое строение» впервые ввёл А. М. Бутлеров 19 сентября 1861 года в докладе «О химическом строении веществ» на химической секции Съезда немецких естествоиспытателей и врачей в Шпейере (опубликованном в том же году на немецком[3] и в следующем — на русском языках[4]). В том же докладе он заложил основы классической теории химического строения. Главные положения этой теории следующие:
атомы в молекулах веществ соединены друг с другом согласно их валентности, порядок распределения связей в молекуле называется химическим строением;
изменение этой последовательности приводит к образованию нового вещества с новыми свойствами;
свойства веществ зависят не только от их качественного и количественного состава, но и от «химического строения», то есть от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильно влияют друг на друга атомы, непосредственно связанные между собой;
атомы в молекулах оказывают влияние друг на друга, и это влияние приводит к химическим изменениям поведения атома;
определить состав и строение химического вещества можно по продуктам химических превращений.
Геометрия молекул
В 1864 году Бутлеров первым объясняет явление изомерии, показав что изомеры — это соединения, обладающие одинаковым элементным составом, но различным химическим строением. В 1874 году возникает стереохимия, или трёхмерная структурная химия в форме постулата Вант-Гоффа о тетраэдрической системе валентностей у атома углерода.
В настоящее время принято различать структурную и пространственную изомерию.
Структурную изомерию подразделяют на изомерию скелета, обусловленную различным порядком связи атомов, образующих скелет молекулы, например в Н-бутане и изобутане, и на изомерию положения одинаковых функциональных групп при одинаковом углеродном скелете молекулы, например в орто- , мета- и пара- изомерах ароматических соединений.
Пространственная изомерия обусловлена существованием стереоизомеров, соединений, имеющих одинаковый порядок связей атомов, но различное пространственное расположение. К видам пространственной изомерии относятся: оптическая изомерия, обусловливающая существование энантиомеров — пары стереоизомеров, представляющих собой зеркальные отражения друг от друга, не совмещаемые в пространстве; диастереомерия, обусловливающая существование изомеров, не являющихся энантиомерами; геометрическая изомерия, обусловливающая цис- и транс- изомеров, свойственных соединениям с двойными связями и малыми циклами.
Электронные интерпретации строения молекул
Строение молекулы H2 по Бору
С открытием в 1897 году электрона (Дж. Томсоном, Э. Вихертом) появились электронные интерпретации строения молекул. Американский физикохимик Г. Льюис в 1912 году предложил электронную теорию химической связи, по которой связь между атомами в молекуле осуществляется обобществлённой электронной парой. Электронная теория химической связи Льюиса стала основой классической теории строения в органической химии, базирующейся на представлении о парной связи между атомами, образованной дублетом электронов. Валентный штрих между символами элементов в молекуле был заменён двумя точками, обозначающими связывающую электронную пару.
В 1913 году датский физик Нильс Бор предложил рассматривать электронную пару в форме вращающегося кольца, плоскость которого перпендикулярна оси молекулы и равноудалена от ядер атомов.
Динамическое равновесие молекулярной системы, по Бору, достигается за счёт баланса сил между силами взаимного отталкивания ядер и силами притяжения ядер к плоскости кольца электронов. Боровская интерпретация строения молекулы учитывала кулоновское отталкивание электронов, в кольце они находятся в диаметрально противоположном положении.
Молекулу метана Нильс Бор описывал следующим образом:
Ядро углерода, заключённое в очень маленькое кольцо из двух электронов, расположено в центре (тетраэдра), а ядро водорода по углам. Химические связи представляют собой четыре двухэлектронных кольца, вращающихся вокруг линий, соединяющих центр с углами[5].
Квантовая природа межатомных сил
Схема диполь-дипольного взаимодействия двух атомов водорода по Лондону
В своём докладе, прочитанном в Королевском колледже в Лондоне в ознаменовании столетия опубликования трудов Дж. Максвелла по электромагнитному излучению, К. Коулсон дал анализ происхождения и сущности межатомных сил, приводящих к образованию молекулы[6]. Коулсон, ссылаясь на работу Лондона, на примере двух атомов водорода показывает «каким образом два нейтральных атома или молекулы могут оказывать притяжение друг к другу на значительном расстоянии».
Ядра A и B двух атомов водорода находятся на расстоянии r друг от друга (рис.). Каждый атом несёт по одном электрону (в P и Q соответственно). Совокупность зарядов +e в A и -e в P приблизительно эквивалентна электрическому диполю, имеющему величину e·AP. Подобным же образом совокупность +e в B и -e в Q приблизительно эквивалентна электрическому диполю e·BQ. Эти два диполя взаимодействуют друг с другом. Общая потенциальная энергия двух диполей, m и m’, находящихся на расстоянии r, равна:
На языке волновой механики это выражение рассматривается как возмущение, действующее на оба атома.
Природа межатомных сил имеет электромагнитный характер и называется диполь-дипольным взаимодействием. Существуют диполь-квадрупольные, квадруполь-квадрупольные и другие взаимодействия, при которых энергия изменяется в зависимости от более высоких степеней 1/r[6].
Распределение электронной плотности в химических соединениях
Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода.
Рис. Модель молекулы водорода и её осевая проекция
Электронная плотность — это плотность вероятности обнаружения электрона в данной точке пространства. Электронная плотность нормирована и, соответственно, указывает на вероятное число электронов в данном элементарном объёме.
Вероятностную (статистическую) интерпретацию волновой функции сформулировал М. Борн в 1926 году как только было опубликована волновое уравнение Шрёдингера. В 1954 году М. Борн удостоен Нобелевской премии по физике с формулировкой «За фундаментальное исследование в области квантовой механики, особенно за статистическую интерпретацию волновой функции».
Расчёт электронной плотности проводят с использованием уравнения Шрёдингера, которое решается аналитически точно для систем, содержащих только один электрон[7].
Получаемая радиальная функция распределения вероятности нахождения электрона в атоме водорода обладает максимумом при α0, как показано на рисунке. Этот наиболее вероятный радиус совпадает с боровским радиусом и принят в качестве атомной единицы для линейных размеров 1 а. е. (бор) = 0,529177·10−10м ≈ 0,529 Å. Более размытое облако плотности вероятности, полученное при решении уравнения Шрёдингера для атома водорода, значительно отличается от боровской модели атома и согласуется с принципами неопределённости Гейзенберга. С учётом статистической интерпретации волновой функции М. Борна и принципа неопределённости Гейзенберга длины диполей AP и BQ взаимодействующих атомов в расчётах Ф. Лондона достаточно размыты. Размыто и электронное кольцо в модели молекулы водорода по Н. Бору до состояния тороидального электронного облака с неопределёнными границами.
Наиболее вероятный радиус электронного кольца (тора) молекулы водорода (re) определяется боровским радиусом (α0) и длиной химической связи (d): re2 = α02 — (d/2)2; re = 0,374 Å.
Благодаря пространственной симметрии дипольный момент молекулы водорода равен нулю, что соответствует её низкой химической активности[8]. Пространственная симметрия электронной плотности сохраняется, если соединяющиеся в молекулу атомы имеют одинаковую энергию ионизации. В этом случае связывающее электронное кольцо располагается на равном расстоянии от каждого из ядер. Если же потенциалы ионизации различны электронная плотность смещается в сторону атома с большим первым потенциалом ионизации[9]. Смещение электронной плотности приводит к асимметрии распределения электрических зарядов в молекуле, молекула становится полярной системой с определённым дипольным моментом.
Приближённые квантовохимические методы расчёта
Поскольку точное решение уравнения Шрёдингера для атомно-молекулярных систем, содержащих два и более электрона, невозможно, предложены приближённые методы расчёта электронной плотности. Все они возникли в 1930-х годах, проделали значительный путь развития и долгое время дополняли друг друга. Наиболее важные из них — теория валентных связей, теория молекулярных орбиталей, теория кристаллического поля, теория функционала плотности.
В рамках теории валентных связей разработана концепция резонанса (Л. Полинг) и родственная ей концепция мезомерии (К. Ингольд). Концепция резонанса рассмотрена на примере молекулярного иона водорода. Л. Полинг показал, что стабильность молекулярного иона водорода:
вызвана резонансом электрона, то есть движением его «взад и вперёд» между двумя ядрами с «резонансной частотой», равной по величине энергии резонанса (50 ккал/моль), делённой на константу Планка h. Для молекулярного иона в основном состоянии эта частота равна 7.1014сек−1[10].
Концепция резонанса дополняла постулаты классической теории химического строения и утверждала, что если для данного соединения классическая теория допускает построение нескольких приемлемых структурных формул, то действительному состоянию молекул этого соединения отвечает не какая-либо одна отдельная формула, а некоторое их сочетание (наложение, резонанс структур)[11].
Мезомерия является теорией электронного строения химических соединений, согласно которой истинное распределение электронной плотности в молекуле является промежуточным между распределениями, представленными несколькими классическими формулами[12].
Обычно рассматривают положительный и отрицательный мезомерные эффекты:
Винилхлорид: +М-эффект
Аллил-катион: −М-эффект
Кампания идеологического вмешательства в теорию химического строения
Кампания началась в 1949 году с публикации статьи В. М. Татевского и М. М. Шахпаронова «Об одной махистской теории в химии и её пропагандистах»[13]. В качестве главного объекта нападения была выбрана теория резонанса Л. Полинга. Было объявлено, что «представления о реальной молекуле как о чём-то среднем между двумя (и более) крайними абстрактными структурами являются буржуазными и поэтому направленными против всего самого „святого“». Были указаны и пропагандисты теории Я. К. Сыркин и М. Е. Дяткина — авторы книги «Химическая связь и строение молекул», в которой нашла отражение теория резонанса.
В воздухе очередной раз запахло инквизицией. В этой тревожной обстановке ведущие химики страны собрались на Всесоюзное совещание по проблемам химического строения (1951 г., Москва). Стенограмма этого совещания — один из наиболее позорных документов, когда-либо созданных коллективными усилиями ученых, хранится в химических библиотеках всего мира, и от этого срама бог весть когда ещё удастся отмыться… До крови не дошло — спасла оттепель, начавшаяся весной следующего года. Я. К. Сыркин и М. Е. Дяткина, подготовленные недавними друзьями и коллегами к выдаче в качестве первых козлов отпущения, уцелели; более того Я. К. Сыркин в дальнейшем стал академиком[14].
Лайнус Полинг удостоен в 1954 году Нобелевской премии по химии «за исследование природы химической связи и её применение для определения структуры сложных соединений».
Однако консенсус в теории химического строения не был достигнут. В. М. Татевский в курсе «Строение молекул» (1977 г.) отмечал:
… полностью выпадают и «висят в воздухе» представления уходящих в прошлое, но всё ещё фигурирующих в литературе так называемых «теории резонанса» и «теории мезомерии», которые не имеют основы ни в классической теории химического строения, ни в законных приложениях классической физики к вопросам строения молекул, ни в квантовой механике[15].
Лишь в 1991 году проведён принципиальный анализ кампании по борьбе с теорией резонанса и было показано, что эта кампания «нанесла серьёзный ущерб престижу советской науки»[16].
Атомы в молекулах
В классической теории химического строения понятие атома в молекуле является изначальным. Интуитивно ясно, что атом в молекуле меняется, меняются и его свойства в зависимости от окружения этого атома, прежде всего ближайшего. Основным показателем является расстояние между атомами в молекуле, определяющее как длину химической связи, так и прочность химической связи.
В квантовой теории понятие атома вторично. Так, по утверждению В. М. Татевского, молекула не состоит из атомов: «С современной точки зрения ясно, что при образовании молекулы сохраняются не атомы, а только ядра атомов и электроны»[17].
Было предпринято множество попыток сохранить понятие атома в молекуле, но, тем не менее, практически всегда они не удовлетворяли последующих исследователей по тем или иным причинам.
Одна из наиболее удачных попыток сохранения классической концепции атома в молекуле принадлежит Р. Бейдеру и его сотрудникам[18]. В рамках этой теории (QTAIM) электронная плотность «задаёт некоторое скалярное поле в трёхмерном пространстве, которое может быть охарактеризовано, например, его совокупностью экстремальных точек, линий и поверхностей, особых точек и т. п.»[19].
Таким образом, в квантовой теории атомов в молекулах Р. Бейдера оказывается возможным физическое обоснование ключевых понятий химии «атом», «молекула», «химическая связь» в терминах топологии функции электронной плотности в трёхмерном пространстве и описание химического строения молекул.
Электронная корреляция и конфигурация молекул
Электронная корреляция (взаимная обусловленность движения всех электронов атомной или молекулярной системы как целого определяется электростатическим отталкиванием электронов и статическим особенностями систем, в частности принципом Паули (фермиевская корреляция). Полный учёт электронной корреляции при расчёте энергии и определении электронной структуры системы достигается методом конфигурационного взаимодействия.
Простая и надёжная система правил для понимания и предсказания конфигурации молекул заложена в теории отталкивания электронных пар, наиболее важное правило которой достаточно эмпирично, хотя имеет квантовомеханическое обоснование, заключающееся в принципе Паули, а именно «электронные пары принимают такое расположение на валентной оболочке атома, при котором они максимально удалены друг от друга». При этом конфигурация молекулы будет определяться числом связывающих и неподелённых электронных пар у центрального атома:
две связывающие электронные пары дают линейную конфигурацию молекулы;
три — конфигурацию правильного треугольника;
четыре — конфигурацию тетраэдра;
пять — тригонально-бипирамидальную конфигурацию;
шесть — конфигурацию октаэдра.
Наличие неподелённых электронных пар у центрального атома приводит к расширению типов конфигураций молекулы[20].
Открытие Н. Бартлеттом в 1962 году первого соединения ксенона положило начало интенсивному развитию химии благородных газов, что привело к получению большого ряда соединений инертных газов и установлению их химического строения.
Оказалось, что все соединения благородных газов имеют обыкновенные ковалентные связи и общепринятые конфигурации молекул. Так, конфигурация молекулы дифторида ксенона линейна: связи ксенон—фтор являются аксиальными и взаимодействуют с тремя неподелёнными электронными парами, находящимися в экваториальных положениях.
В молекуле тетрафторида ксенона осуществляется конфигурация с плоским квадратным расположением лигандов. Неподелённые электронные пары ксенона максимально удалены друг от друга и расположены по разные стороны от этого квадрата.
Структура молекулы гексафторида ксенона представляет собой искажённый октаэдр. «Отклонение атомов фтора от вершин правильного октаэдра указывает на то, что неподелённая электронная пара занимает положение над центром одной из граней октаэдра, а атомы фтора, расположенные в углах этой грани, вынуждены раздвинуться»[20].
Открытие Бартлетта показало ошибочность популярного в то время правила октета,[как?] согласно которому при образовании молекул атомы удовлетворяют свою потребность в достижении 8-электронной валентной оболочки, подобной электронной конфигурации благородных газов за счёт попарного обобществления своих валентных электронов. Оказалось, что атомы благородных газов, имея полностью заполненную валентную оболочку, могут вступать в химические реакции и участвовать в химическом строении молекул.
Строение электронодефицитных соединений
Структурная проблема, связанная с электронодефицитными соединениями, довольно сложна. Фундаментальная трудность заключалась в том, что в молекулах нет достаточного числа валентных электронов для того, чтобы связать все атомы обычными двухэлектронными связями. Так, например, в молекуле диборана имеется двенадцать валентных электронов, все двенадцать нужны для образования ковалентных связей шести атомов водорода с бором, так что для связи атомов бора между собой электронов не остаётся. Сам Полинг допускал, что в диборане функционируют одноэлектронные связи, а молекула в основном состоянии резонирует между семью структурами льюисовского типа, а также между многочисленными структурами, содержащими одноэлектронные связи.
Однако заслуженное признание получили исследования природы химической связи в бороводородах, выполненные американским физикохимиком У. Липскомбом. В его интерпретации в диборане имеют место четыре двух- и две трёхцентровые связи.
Четыре концевые двухцентровые двухэлектронные связи HB лежат в одной плоскости. Два же центральных атома водорода расположены симметрично над этой плоскостью и под нею и объединены с атомами бора двумя трёхцентровыми связями.
В 1976 году Липскомб удостоен Нобелевской премии по химии с формулировкой «за исследование структуры боранов (боргидритов), проясняющих проблему химических связей».
Особенности строения сэндвичевых соединений
Молекула ферроцена
Дальнейшее развитие теории химического строения связано с открытием и установлением структуры ферроцена[21]. Оказалось, что при взаимодействии хлорида двухвалентного железа и циклопентадиена вместо ожидаемого соединения с двумя химическими связями железо-углерод образуется сэндвичеобразное соединение, в котором все 10 атомов углерода соединены с атомом железа.
Рентгеноструктурными исследованиями Э. Фишера установлено, что все атомы углерода в молекуле ферроцена структурно и химически эквивалентны. Атом металла взаимодействует не с одним или двумя конкретными атомами углерода, а со всеми атомами углерода двух циклопентадиенильных колец сразу. Атом металла как бы завис в пространстве между двумя циклами, представленных параллельными правильными пятиугольниками.
В настоящее время получены металлоцены — бициклопентадиенильные соединения переходных металлов для большинства d-элементов.
Сэндвичевые соединения образуются также с использованием в качестве органического фрагмента бензола или циклооктатетраена. Так, например, в ураноцене (см. рисунок) атом урана удерживает два восьмичленных кольца. Все 16 связей уран—углерод в ураноцене идентичны.
В 1973 году Э. Фишер и Дж. Уилкинсон удостоены Нобелевской премии по химии с формулировкой «За новаторскую, проделанную независимо друг от друга, работу в области металлорганических, так называемых сэндвичевых соединений».
Считается, что в сэндвичевых соединениях реализуется многоцентровая связь, тип химической связи, в которой в отличие от двухцентровой двухэлектронной связи, связывающие электронные пары распределены в пространстве трех или большего числа атомных центров молекулы, иона, радикала.[22]
Примечания
↑ 12 Совещание по теории химического строения в органической химии. Успехи физических наук.. — 1951, октябрь. — Т. XLV, вып. 2. — С. 277—392.
↑ Химический энциклопедический словарь / гл. ред. И. Л. Кнунянц. — Сов. энциклопедия, 1983. — С. 652. — 792 с.
↑ Butlerow A. Einiges über die chemische Structur der Körper. (Vorgetragen in der chemischen Section der 36. Versammlung deutscher Naturforscher und Aerzte zu Speyer am 19.Septbr.) (нем.) // Zeitschrift für Chemie und Pharmacie : magazin. — 1861. — Bd. 4. — S. 549—560.
↑ Бутлеров А. О химическом строении веществ // Учёные записки Казанского университета (отд. физ.-мат. и мед. наук). Вып.1, отд.1. — 1862. — С. 1—11.
↑ Бор Н. Избранные научные труды. — М.: «Наука», 1970. — Т. 1. — С. 147. — 584 с.
↑ 123Коулсон К. Успехи физических наук (статья «Межатомные силы — от Максвелла до Шрёдингера»). — 1963, ноябрь. — Т. LXXXI, вып.3. — С. 545-556.
↑ Шусторович Е. М. Химическая связь. Сущность и проблемы.. — М.: «Наука», 1973. — С. 6. — 232 с.
↑ Потапов А. А. Ренессанс классического атома. — М.: «Наука», 2011. — С. 352. — 444 с.
↑ Ганкин В. Ю., Ганкин Ю. В. Как образуется химическая связь и протекают химические реакции. — М.: «Граница». — С. 65. — 320 с.
↑ Паулинг Л. Природа химической связи. — М.-Л.: Химическая литература, 1947. — С. 25—26. — 440 с.
↑ Новая иллюстрированная энциклопедия. Пр-Ро. Кн.15. — М.: Большая российская энциклопедия, 2002. — С. 169. — 255 с.
↑ Новая иллюстрированная энциклопедия. Ма-Мо. Кн.11. — М.: Большая российская энциклопедия, 2002. — С. 141. — 255 с.
↑ Вопросы философии. — 1949. — № 3. — С. 176—192.
↑ Охлобыстин О. Ю. Жизнь и смерть химических идей. Очерки по истории теоретической химии.. — М.: Наука, 1989. — С. 184. — 190 с.
↑ Татевский В. М. Строение молекул.. — М.: Химия, 1977. — С. 10. — 512 с.
↑ Сонин А. С. Печальный юбилей одной кампании. (рус.) // Вестник РАН. — 1991. — Т. 61, № 8. — С. 96—107. Архивировано 5 мая 2010 года.
↑ Татевский В. М. Квантовая механика и теория строения молекул. — М.: Издательство МГУ, 1965. — С. 17. — 162 с.
↑ Бейдер Р. Атомы в молекулах. Квантовая теория. — М.: Мир, 2001. — 532 с.
↑ Степанов Н. Ф. Квантовая механика и квантовая химия. — М.: Мир, 2001. — С. 487. — 519 с.
↑ 12Р. Гиллеспи. Геометрия молекул. — М.: Мир, 1975. — 278 с.
↑ Несмеянов А.Н. Ферроцен и родственные соединения. Избранные труды 1969 — 1979. — М.: Советская энциклопедия, 1982.
↑
Химический Энциклопедический словарь. — М.: Советская энциклопедия, 1983. — 792 с.
ru-wiki.org
XuMuK.ru — Теория химического строения
К шестидесятым годам прошлого столетия в органической химии накопился огромный фактический материал, который требовал объяснения. На фоне беспрерывного накопления экспериментальных фактов особенно остро проявлялась недостаточность теоретических представлений органической химии. Теория отставала от практики, от эксперимента. Такое отставание болезненно отражалось на ходе экспериментальных исследований в лабораториях; химики проводили свои исследования в значительной мере наугад, вслепую, зачастую не понимая природы синтезированных ими веществ и сути реакций, которые приводили к их образованию. Так, например, англичанин В. Перкин старш., синтезировавший в1856 г. краситель мовеин окислением нечистого анилина, совершенно не представлял себе механизма открытой им реакции; к тому же он вовсе не ставил перед собой задачу синтеза красителя, а пытался получить хинин. Органическая химия, по меткому выражению Вёлера, напоминала дремучий лес, полный чудесных вещей, огромную чащу без выхода, без конца.
Насущные задачи органической химии требовали разрешения основного вопроса: являются ли молекулы беспорядочным нагромождением атомов, удерживаемых силами притяжения, или же молекулы представляют собой частицы с определенным строением, которое можно установить, исследуя свойства вещества. Теория типов Жерара, с теми или иными оговорками признававшаяся большинством химиков того времени, отказывалась на основании изучения химических свойств решать вопрос о строении молекул. А между тем в органической химии к этому времени уже накопились факты и обобщения, которые могли служить основой для решения этого вопроса. Так, например, теория радикалов дала органической химии чрезвычайно важное обобщение, заключавшееся в том, что при химических реакциях некоторые группы атомов в неизменном виде переходят из молекул исходных веществ в молекулы, образующиеся при этих реакциях. Теория типов, со своей стороны, в значительной мере способствовала изучению наиболее изменчивых частей молекул и причин этой изменчивости.
Необычайно большое значение имело открытие валентности элементов.
Исследуя состав металлоорганических соединений, Франк-ланд (1853) нашел, что каждый металл дает соединения со строго определенным числом радикалов; это число и представляет собой валентность данного металла. Ниже приведены простейшие металлоорганические соединения одно-, двух-, трех- ичетырехвалентных металлов:
После открытия Франкланда стало ясно, что атомы могут соединяться в молекулы только в отношениях, определяемых валентностью атомов. В частности, было установлено, что углерод четырехвалентен (Кекуле, Кольбе).
Открытие валентности непосредственно подводило кмысли, что молекулы имеют определенное строение. Однако вопросы о путях установления строения молекул, а также о зависимости свойств вещества от строения его молекул оставались открытыми.
Новая эпоха в органической химии наступила с возникновением теории химического строения. Главная роль в создании.
обосновании и подтверждении теории строения принадлежит знаменитому русскому ученому Александру Михайловичу Бутлерову, хотя кроме него элементы этой теории начали разрабатывать А. Купер (1831—1892) в Англии и А. Кекуле (1829— 1896) в Германии.
В 1858 г. Купер опубликовал на трех языках (английском, французском и немецком) статью «О новой химической теории», где он отбрасывает теорию типов и высказывает точку зрения, согласно которой все особенности органических веществ могут быть объяснены, если учитывать только два свойства атомов: «избирательное сродство» (связь атомов) и «степень сродства» (валентность атомов).
Купер писал: «С моей точки зрения этих двух свойств достаточно для объяснения всего того, что характерно для органической химии: именно это я докажу ниже… В молекуле, состоящей из трех, четырех, пяти и т. д. атомов углерода и эквивалентного количества водорода, кислорода и др., последние могут быть заменены другими элементами, в то время как углерод образует взаимно-связанный узел. Это означает, что один углерод связан с другим углеродом. Такое свойство придает углероду, так сказать, своеобразную физиономию и дает возможность понять непонятный до этого факт наслоения атомов углерода в органических соединениях».
Придя таким образом к важному представлению о цепи углеродных атомов, Купер выражает далее свои взгляды в формулах, которые, по его замыслу, должны дать картину строения соединений. В качестве примера его формул, которые были первыми конституционными формулами, можно привести следующие:
Из этих примеров видно, что Куперу удалось удивительно правильно передать конституцию этих соединений, а также некоторых более сложных и в то время мало исследованных (винная и виноградная кислоты).
Однако все эти формулы были лишены опытного обоснования. Купер совершенно не ставил вопроса о возможности их экспериментальной проверки. Его формулы, как легко видеть, были основаны на формальной интерпретации понятий валентности и связи атомов, а отчасти даже на интуиции. Естественно, что при таком подходе невозможно избежать ошибок. Так, например, формулы глицерина, глицериновой кислоты и щавелевой кислоты, данные Купером, уже неверны:
Таким образом, взгляды Купера, развитые им в его талантливой, интересной работе, не носят характера строгой теории.
Другая попытка изображения органических соединений конституционными формулами была сделана в 1861 г. Лошмидтом. При построении своих формул Лошмидт рассматривал атомы как мельчайшие материальные частицы, подвергающиеся действию сил притяжения и отталкивания. Эти силы при сближении атомов уравновешиваются, и различные атомы удерживаются друг около друга в некотором равновесном положении. Сферы действия атомных сил Ло-шмидт условно обозначал кружками (например, атомы углерода и водорода — простыми кружками, кислорода—двойными, азота—тройными).
Формулы Лошмидта имели следующий вид:
Не пытаясь составить какое-либо представление о способе связи шести углеродных атомов в молекуле бензола, Лошмидт обозначал бензол символом
В отличие от Купера, Лошмидт при выборе формул, кроме валентности («поллентности» по его выражению), иногда руководствовался и химическими свойствами. Однако в целом метод вывода формул Лошмидта был абстрактным, а зачастую просто необоснованным. Так, не опираясь на химические данные, Лошмидт пытался вывести формулы таких сложных веществ, как индиго, мочевая кислота и т. п.
Естественно, что эти формулы оказались ошибочными.
Несмотря на то, что многие из предложенных Лошмидтом формул органических соединений оказались удачными, работа его осталась почти не замеченной химиками того времени и не оказала сколько-нибудь существенного влияния на развитие теории органической химии.
Большой вклад в создание структурной теории внес знаменитый немецкий химик Кекуле. Он установил четырехвалент-ность углерода, ввел тип метана, предложил известную формулу бензола и, что особенно важно, правильно сформулировал одну из основных задач органической химии того времени.
В статье «О конституции и превращениях химических соединений и о химической природе углерода» в 1858 г. Кекуле писал: «Я считаю, что в настоящее время главной задачей химии не является обнаружение атомных групп, которые, вследствие некоторых своих свойств, могут быть рассматриваемы как радикалы, и причисление соединений к некоторым типам, которые едва ли при этом имеют иное значение, кроме как образца формулы. Напротив, я полагаю, что необходимо распространить размышление и на строение самих радикалов; из природы элементов должна быть выведена как природа радикалов, так и их соединений». Исходным пунктом для этого Кекуле считал «основность элемента» (валентность), а в отношении органических соединений прежде всего — природу углерода. Кекуле высказал также ряд других верных мыслей о связи атомов, выражая ее графическими формулами. Однако своим формулам Кекуле не придавал значения формул строения, он стремился выразить ими только реакционную способность. Так, он писал: «Рациональные формулы имеют своей целью дать определенное представление о химической природе соединения, следовательно, о его метаморфозах и отношениях, в которых оно находится к другим телам… При этом, естественно, необходимо иметь в виду, что рациональные формулы — это лишь формулы превращений, а не конституционные формулы, что они являются лишь средством выражения для метаморфоз тел и результатов сравнения различных веществ между собою; что они ни в коем случае не должны выражать конституцию, т. е. расположение атомов в соответствующем соединении».
Установление истинного строения молекул Кекуле также считал задачей химии, но, по его мнению, это может быть достигнуто не путем изучения химических превращений, а только сразнительным исследованием физических свойств соединений. Таким образом, и в этом вопросе Кекуле стоял на позиции Же-рара.
Как и другие сторонники теории типов, Кекуле изображал вещество несколькими типическими формулами. Так, например, чтобы передать известные тогда химические свойства уксусной кислоты, Кекуле предлагал изображать ее восемью формулами. Таким образом, хотя взгляды Кекуле и были близки к новым структурным воззрениям, хотя Кекуле и внес существенный вклад в развитие теории химического строения, он так и не смог полностью освободиться от представлений теории типов.
А. М. Бутлеров выступил против положения теории типов о невозможности установления строения молекул химическим путем; он показал, что в молекуле имеется определенная последовательность химической связи атомов (химическое строение). Далее Бутлеров доказал, что строение молекулы можно установить, исследуя химические свойства вещества, и, наоборот, зная строение, можно предвидеть многие свойства соединения. Бутлеров не только обосновал это положение уже имевшимся фактическим материалом, но и предсказал на его основе возможность существования новых веществ, которые впоследствии были открыты им и другими химиками.
Основная идея теории А. М. Бутлерова сформулирована им в 1861 г. в статье «О химическом строении веществ». Он писал: «Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определенным количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которого химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу».
Основой теории Бутлерова является идея о порядке химического взаимодействия атомов в молекуле. Этот порядок химического взаимодействия не включает представления о механизме химической связи и физическом расположении атомов. Эта важная особенность теории химического строения позволяет всегда опираться на нее при построении физической модели молекулы.
Установив понятие химического строения, А. М. Бутлеров дает новое определение природы вещества: «химическая натура сложной частицы определяется натурой элементарных составных частей, количеством их и химическим строением».
Таким образом, А. М. Бутлеров первый установил, что каждая молекула имеет определенное химическое строение, что строение определяет свойства вещества и что, изучая химические превращения вещества, можно установить его строение.
Взгляды А. М. Бутлерова на значение химических структурных формул вытекают из основных положений его теории. Бутлеров считал, что эти формулы должны быть не «типическими», «реакционными», а конституционными. В этом смысле для каждого вещества возможна лишь одна рациональная формула, на основании которой можно судить о химических свойствах.
Что же касается способа написания структурных формул, то Бутлеров справедливо считал этот вопрос второстепенным: «Помня, что дело не в форме, а в сущности, в понятии, в идее,— и принимая во внимание, что формулами, обозначающими изомерию, логически-необходимо выражать настоящее частицы, т. е. некоторые химические отношения, в ней существующие,— не трудно притти к убеждению, что всякий способ писания может быть хорош, лишь бы только он с удобством выражал эти отношения. Весьма естественно даже употреблять разные способы, предпочитая тот, который является более выразительным для данного случая. Например, этан С2Н6 почти совершенно безразлично может быть изображен:
…Однако же, при недостаточно-определенном понимании, иной способ писания может привести к недоумениям».
С возникновением теории химического строения органическая химия вышла из лабиринта типических формул; были показаны пути к познанию внутреннего строения молекул; появилась теоретическая основа для понимания химических процессов, для предсказания новых путей синтеза органических соединений. С самого момента своего возникновения теория химического строения дала возможность химикам проводить экспериментальные исследования направленно, целеустремленно.
Замечательным успехом теории химического строения явилось объяснение явления изомерии, открытого в первой четверти прошлого столетия.
Как известно, в конце XVIII века был установлен закон постоянства состава, согласно которому каждое данное вещество имеет определенный, постоянный состав. В течение нескольких десятков лет этому закону придавалось и обратное значение, т. е. считалось,что данным определенным составом обладает лишь одно определенное вещество. Неправильность последнего положения была показана в результате исследования ряда органических веществ. В 1823 г. Либих, исследовав серебряную соль гремучей кислоты, установил, что ее состав (AgCNO) тождествен составу полученного в 1822 г. Вёлером и резко отличного от нее изоциановокислого серебра.
Этот замечательный факт недолго оставался единичным; вскоре были обнаружены многие другие вещества, обладающие одинаковым составом, но разными свойствами. Открытое явление с 1830 г. начали называть изомерией (от греч. — составленный из одинаковых частей), а вещества с одинаковым составом — изомерами. Попытки объяснить изомерию теорией радикалов и теорией типов были односторонними (как и сами эти теории), а потому не давали удовлетворительных результатов. Фактически в течение почти четырех десятилетий явление изомерии не находило теоретического объяснения.
Такое объяснение стало возможным только после создания теории химического строения, согласно которой природа вещест-ва определяется не только характером и числом атомов, входящих в состав молекулы, но и ее структурой, химическим строением. Отсюда с несомненной очевидностью вытекает возможность существования веществ, имеющих одинаковый состав и молекулярный вес и, тем не менее, совершенно различных вследствие неодинакового химического строения. Таким образом, различие в химическом строении явилось естественным и простым объяснением явления изомерии.
Следует отметить, что А. М. Бутлеров открыл и впервые объяснил также явление динамической изомерии, заключающееся в том, что два или несколько изомеров в определенных условиях легко переходят друг в друга (явление это в настоящее время носит название таутомерия). Проблема изомерии в целом явилась серьезным испытанием для бутлеровской теории и была ею блестяще решена.
Важной особенностью учения А. М. Бутлерова является то что он отнюдь не считал молекулу каким-то неподвижным образованием, в котором отдельные атомы связаны в мертвую, безжизненную конструкцию. По этому поводу он писал:
«…в настоящее время мы смотрим на химическое соединение не как на что-либо мертвое, неподвижное; мы принимаем, напротив, что оно одарено постоянным движением, заключенным в его самых мельчайших частичках, частные взаимные отношения которых подлежат постоянным переменам, суммируясь при этом в некоторый постоянный средний результат. Мы можем иметь здесь и постоянные изменения в химических частицах, составляющих массу веществ, но все это сводится к известному среднему состоянию самой массы. Словом, вообще мы имеем всегда перед собою состояние известного подвижного равновесия. С этой динамической точки зрения на натуру химического соединения и на химические реакции, мы ясно объясняем такие явления, которые, с прежней точки зрения, были совсем непонятны. Стоит указать, например, на диссоциацию,— на то, как легко мы теперь объясняем обратные реакции и т. п.».
Сформулированные А. М. Бутлеровым ясные, неопровержимые положения теории химического строения в короткий срок обеспечили ей всеобщее признание. Однако вместе с тем появилась тенденция замолчать заслуги А. М. Бутлерова и представить творцами теории строения только Кекуле и Купера.
Уже несколько лет спустя после создания теории строения А. М. Бутлерову пришлось выступить в защиту своего приоритета, так как некоторые зарубежные химики, вначале не признававшие и даже не понимавшие его теории, впоследствии пытались приписать честь создания основных положений этой теории себе.
Решающую роль А. М. Бутлерова в создании теории химического строения ярко подчеркнул в 1868 г. великий русский ученый Д. И. Менделеев, рекомендуя А. М. Бутлерова в Петербургский университет. Менделеев писал, что Бутлеров «…вновь стремится, путем изучения химических превращений, проникнуть в самую глубь связей, скрепляющих разнородные элементы в одно целое, придает каждому из них прирожденную способность вступать в известное число соединений, а различие свойств приписывает различному способу связи элементов. Никто не проводил этих мыслей столь последовательно, как он, хотя они и проглядывали ранее… Для проведения того же способа воззрения через зсе классы органических соединений Бутлеров издал в 1864 г. книгу: «Введение к полному изучению органической химии», в прошлом году переведенную на немецкий язык Бутлеров чтениями и увлекательностью идей образовал вокруг себя в Казани школу химиков, работающих в его направлении. Имена Марковникова, Мясникова, Попова, двух Зайцевых, Моргунова и некоторых других успели получить известность по многим открытиям, сделанным преимущественно благодаря самостоятельности бутлерозского направления. Могу лично засвидетельствовать, что такие ученые Франции и Германии, как Вюрц и Кольбе, считают Бутлерова одним из влиятельнейших в наше время двигателей теоретического направления химии».
А. М. Бутлеров справедливо считал, что теория химического строения будет развиваться по мере накопления нового фактического материала. Он писал: «…не могу не заметить, что те заключения, к которым ведет принцип химического строения, оказываются в тысячах случаев согласными с фактами. Как во всякой теории, и здесь, конечно, есть недостатки, несовершенства, — встречаются факты, которые не отвечают строго понятию о химическом строении. Разумеется, следует желать в особенности размножения таких именно фактов; факты, не объясняемые существующими теориями, наиболее дороги для науки, от их разработки следует по преимуществу ожидать ее развития в ближайшем будущем»).
Теория химического строения создавалась А. М. Бутлеровым в середине XIX века, в период, когда в России вырастали но-зые буржуазные общественно-экономические отношения и рост производительных сил привел к мощному развитию естествознания. В этот период И. М. Сеченов и затем И. П. Павлов создают материалистическое учение о высшей нервной деятельности человека и животных, К. А. Тимирязев и несколько позднее И. В. Мичурин закладывают начало нового этапа в развитии биологии, Д. И. Менделеев открывает важнейший закон природы — периодический закон, обобщивший все имевшиеся в то время знания о химических элементах. Н. И. Лобачевский открывает новую область математики.
Теория химического строения создала возможность научной систематизации фактического материала органической химии, объяснила ее важнейшие закономерности и дала ключ к предсказанию новых фактов. Она явилась научной основой для создания современной органической химии.
формировать понятия о сущности теории
химического строения органических веществ,
опираясь на знания учащихся об электронном
строении атомов элементов, их положении в
Периодической системе Д.И. Менделеева, о
степени окисления, природе химической связи и о
других главнейших теоретических положениях:
последовательность расположения атомов
углерода в цепи,
взаимное влияние атомов в молекуле,
зависимость свойств органических веществ от
структуры молекул;
сформировать представление о ходе развития
теорий в органической химии;
усвоить понятия: изомеры и изомерия;
разъяснить смысл структурных формул
орг.веществ и их преимуществ перед
молекулярными;
показать необходимость и предпосылки создания
теории химического строения;
продолжить формирование навыков составления
конспекта.
тренировать внимание учащихся при восприятии
большого по объему материала;
выробатывать умения анализировать информацию и
выделять наиболее важный материал.
Воспитательные:
с целью патриотического и интернационального
воспитания привести учащимся исторические
сведения о жизни и деятельности ученых.
ХОД УРОКА
1. Организацонная часть
– Приветствие
– Подготовка учащихся к уроку
– Получение сведений об отсутствующих.
2. Изучение нового
План лекции: <Приложение 1.
Слайд 2>
I. Доструктурные теории:
– витализм;
– теория радикалов;
– теория типов.
II. Краткая справка о состоянии химической науки к
60-м годам XIX столетия. Условия создания теории
химического строения веществ:
– необходимость создания теории;
– предпосылки теории химического строения.
III. Сущность теории химического строения
органических веществ А.М. Бутлерова. Понятие об
изомерии и изомерах.
IV. Значение теории химического строения
органических веществ А.М. Бутлерова и ее
развитие.
3. Задание на дом: конспект, п. 2.
4. Лекция
I. Знания об органических веществах
накапливались постепенно еще с глубокой
древности, но как самостоятельная наука
органическая химия возникла лишь в начале XIX
века. Оформление самостоятельности орг.химии
связано с именем шведского ученого Я. Берцелиуса
<Приложение 1. Слайд
3>. В 1808-1812 г.г. он издал свое большое руководство
по химии, в котором первоначально намеревался
рассмотреть наряду с минеральными также и
вещества животного и растительного
происхождения. Но часть учебника, посвященная
орг.веществам, появилась лишь в 1827 г.
Самое существенное различие между веществами
неорганическими и органическими Я. Берцелиус
видел в том, что первые могут быть получены в
лабораториях синтетическим путем, в то время как
вторые якобы образуются лишь в живых организмах
под действием некой «жизненной силы» –
химического синонима «души», «духа»,
«божественного происхождения» живых
организмов и составляющих их органических
веществ.
Теория, объяснявшая образование орг.соединений
вмешательством «жизненной силы», получила
название витализма. В течение
некоторого времени она пользовалась
популярностью. В лаборатории удавалось
синтезировать лишь самые простые
углеродсодержащие вещества, такие как
углекислый газ – СО2, карбид кальция – CaC2,
цианид калия – KCN.
Только в 1828 г. немецкий ученый Вёлер <Приложение
1. Слайд 4> сумел получить органическое
вещество мочевину из неорганической соли –
цианата аммония – NH4CNO.
NH4CNO ––t–> CO(NH2)2
В 1854 г. французский ученый Бертло <Приложение
1. Слайд 5>получил триглицерид. Это и
повлекло за собой необходимость изменения
определения органической химии.
Ученые пытались на основании состава и свойств
разгадать природу молекул органических веществ,
стремились создать систему, которая позволила бы
связать воедино разрозненные факты,
накопившиеся к началу XIX века.
Первая попытка создания теории, стремившейся
обобщить имевшиеся об орг.веществах данные,
связана с именем французского химика Ж.Дюма <Приложение 1. Слайд 6>.
Это была попытка рассмотреть с единой точки
зрения довольно большую группу орг.соединений,
которые сегодня мы называли бы производными
этилена. Орг.соединения оказывались
производными некоторого радикала C2H4
– этерина:
C2H4 * HCl – хлористый этил (солянокислый
этерин)
Заложенная в этой теории идея – подход к
орг.веществу как состоящему из 2-х частей – легла
в последствии в основу, более широкой теории
радикалов (Я. Берцелиус, Ю.Либих, Ф. Велер). Эта
теория основана на представлении о
«дуалистическом строении» веществ. Я.
Берцелиус писал: «каждое орг.вещество состоит
из 2-х составных частей, несущих противоположный
электрический заряд». Одной из этих составных
частей, а именно частью электроотрицательной,
Я.Берцелиус считал кислород, остальная же часть,
собственно органическая, должна была составлять
электроположительный радикал.
Основные положения теории радикалов:<Приложение 1. Слайд 7>
– в состав органических веществ входят
радикалы, несущие на себе положительный заряд;
– радикалы всегда постоянны, не подвергаются
изменениям, они без изменений переходят из одной
молекулы в другую;
– радикалы могут существовать в свободном
виде.
Постепенно в науке накапливались факты,
противоречащие теории радикалов. Так Ж.Дюма
провел замещение водорода хлором в
углеводородных радикалах. Ученым, приверженцам
теории радикалов, казалось невероятным, чтобы
хлор, заряженный отрицательно, играл в
соединениях роль водорода, заряженного
положительно. В 1834 г. Ж. Дюма получил задание
расследовать неприятное происшествие во время
бала во дворце французского короля: свечи при
горении выделяли удушливый дым. Ж.Дюма установил,
что воск, из которого делались свечи, фабрикант
для отбелки обрабатывал хлором. При этом хлор
входил в молекулу воска, заменяя часть
содержавшегося в ней водорода. Удушливые пары,
перепугавшие королевских гостей, оказались
хлороводородом (HCl). В дальнейшем Ж.Дюма
получил трихлоруксусную кислоту из уксусной.
Таким образом, электроположительный водород
заменялся крайне электроотрицательным
элементом хлором, а свойства соединения при этом
почти не менялись. Тогда Ж.Дюма сделал вывод, что
на место дуалистического подхода должен стать
подход к орг.соединению как единому целому.
Теория радикалов была постепенно отвергнута,
однако она оставила глубокий след в
органической химии: <Приложение
1. Слайд 8>
– понятие «радикал» прочно вошло в химию;
– верным оказалось утверждение о
возможности существования радикалов в свободном
виде, о переходе в огромном числе реакций
определенных групп атомов из одного
соединения в другое.
В 40-х г.г. XIXв. Было положено начало учению о
гомологии, позволившему выяснить некоторые
отношения между составом и свойствами
соединений. Выявлены гомологические ряды,
гомологическая разность, что позволило
классифицировать органические вещества.
Классификация орг.веществ на основе гомологии
привела к возникновению теории типов (40-50-е годы
XIX в., Ш. Жерар, А.Кекуле и др.) <Приложение
1. Слайд 9>
Сущность теории типов <Приложение
1. Слайд 10>
– в основу теории положена аналогия в реакциях
между органическими и некоторыми
неорганическими веществами, принятыми в
качестве типов ( типы: водород, вода, аммиак,
хлороводород и др.). Замещая в типе вещества атомы
водорода на другие группы атомов, ученые
предсказали различные производные. Например,
замещение атома водорода в молекуле воды на
радикал метил приводит к возникновению молекулы
спирта. Замещение двух атомов водорода – к
появлению молекулы простого эфира <Приложение
1. Слайд 11>
Ш. Жерар прямо говорил в связи с этим, что
формула вещества – это только сокращенная
запись его реакций.
Все орг. вещества считали производными
простейших неорганических веществ – водорода ,
хлороводорода, воды, аммиака <Приложение
1. Слайд 12>
<Приложение 1. Слайд
13>
– молекулы органических веществ
представляют собой систему, состоящую из атомов,
порядок соединения которых неизвестен; на
свойства соединений влияет совокупность всех
атомов молекулы;
– невозможно познать строение вещества, так как
молекулы в процессе реакции изменяются.
Формула вещества отражает не строение, а
реакции, в которые данное вещество. Для
каждого вещества можно написать столько
рациональных формул, сколько различных видов
превращений может испытывать вещество.
Теория типов допускала множественность
«рациональных формул» для веществ в зависимости
от того какие реакции хотят этими формулами
выразить.
Теория типов сыграла большую роль в
развитии органической химии <Приложение
1. Слайд 14>
– позволила предсказать и открыть ряд
веществ;
– оказала положительное влияние на развитие
учения о валентности;
– обратила внимание на изучение химических
превращений органических соединений, что
позволило глубже изучить свойства веществ, а
также свойства предсказываемых соединений;
– создала совершенную для того времени
систематизацию органических соединений.
Не следует забывать, что в действительности
теории возникали и сменяли друг друга не
последовательно, а существовали одновременно.
Химики нередко плохо понимали друг друга. Ф.Вёлер
в 1835 г. говорил, что «органическая химия в
настоящее время может кого угодно свести с ума.
Она представляется мне дремучим лесом полным
чудесных вещей, огромной чащей без выхода, без
конца, куда не осмеливаешься проникнуть…».
Ни одна из этих теорий не стала теорией
органической химии в полном смысле слова.
Главная причина несостоятельности этих
представлений в их идеалистической сущности:
внутреннее строение молекул считалось
принципиально непознаваемым, а любые
рассуждения о нем – шарлатанством.
Нужна была новая теория, которая бы стояла на
материалистических позициях. Такой теорией
явилась теория химического строения А.М.
Бутлерова <Приложение 1.
Слайды 15, 16>, которая создана в 1861 г. Все
рациональное и ценное, что было в теориях
радикалов и типов, было в дальнейшем
ассимилировано теорией химического строения.
Необходимость появления теории
диктовалась: <Приложение 1.
Слайд 17>
– возросшими требованиями промышленности к
органической химии. Необходимо было
обеспечить текстильную промышленность
красителями. В целях развития пищевой
промышленности требовалось усовершенствовать
методы переработки сельскохозяйственных
продуктов.
В связи с этими задачами начали разрабатываться
новые методы синтеза органических веществ.
Однако у ученых возникли серьезные
затруднения по научному обоснованию этих
синтезов. Так, например, нельзя было объяснить
валентность углерода в соединениях с помощью
старой теории.
Углерод нам известен как элемент 4-х валентный
(Это было доказано экспериментально). Но здесь он
как будто только в метане CH4 сохраняет эту
валентность. В этане C2H6 если
следовать нашим представлениям, углерод д.б.
3-валентным, а в пропане C3H8 – дробную
валентность. (А мы знаем, что валентность должна
быть выражена только целыми числами).
Какова же валентность углерода в органических
соединениях?
Было непонятно, почему существуют вещества с
одинаковым составом, но различными свойствами: С6H12O6
– молекулярная формула глюкозы, но такая же
формула и фруктозы (сахаристого вещества –
составной части мёда).
Доструктурные теории не могли объяснить
многообразие органических веществ. (Почему
углерод и водород – два элемента, – могут
образовывать такое большое число различных
соединений?).
Необходимо было систематизировать имеющиеся
знания с единой точки зрения и разработать
единую химическую символику.
Научно обоснованный ответ на эти вопросы
дала теория химического строения органических
соединений, созданная русским ученым А.М.
Бутлеровым.
Основными предпосылками,
подготовившими почву для возникновения
теории химического строения были <Приложение
1. Слайд 18>
– учение о валентности. В 1853 г. Э. Франкланд
ввел понятие о валентности, установил
валентность для ряда металлов, исследуя
металлоорганические соединения. Постепенно
понятие валентности было распространено на
многие элементы.
В 1858 г. А. Кекуле предложил считать атом
углерода четырехвалентным.
Важным открытием для органической химии
явилась гипотеза о способности атомов углерода к
образованию цепей (А. Кекуле, А. Купер).
Одной из предпосылок была выработка
правильного представления об атомах и молекулах.
До 2-й половины 50-х г.г. XIXв. Не было общепризнанных
критериев для определения понятий: «атом»,
«молекула», «атомная масса», «молекулярная
масса». Только на международном конгрессе
химиков в Карлсруэ (1860 г.) были четко определены
эти понятия, что предопределило развитие
теории валентности, возникновение теории
химического строения.
Основные положения теории химического
строения А.М. Бутлерова (1861 г.)
А.М. Бутлеров сформулировал важнейшие идеи
теории строения органических соединений в виде
основных положений, которые можно разделить на 4
группы.<Приложение 1.
Слайд 19>
1. Все атомы, образующие молекулы
органических веществ, связаны в определенной
последовательности согласно их валентности (т.е.
молекула имеет строение).
<Приложение 1.
Слайды 19, 20>
В соответствии с этими представлениями
валентность элементов условно изображают
черточками, например, в метане CH4. <Приложение 1. Слайд 20>>
Такое схематичное изображение строения
молекул называют формулами строения и
структурными формулами. Основываясь на
положениях о 4-х валентности углерода и
способности его атомов образовывать цепи и
циклы, структурные формулы орг.веществ можно
изобразить так: <Приложение
1. Слайд 20>
ирония, сатира, сарказм, как способ создания характера героя
Комедия представляет собой вид драмы, который отличается сатирическим и юмористическим подтекстом, через который происходит развязка борьбы антагонистических персонажей либо же действенного конфликта.
Комедия как драматургический жанр
Комедия, как драматургический жанр, возникла еще в Древней Греции. Аристотель определял термин «комедия» как демонстрация людям их же пороков в юмористическом виде. В драматургии различают два вида комедий: комедия положений и комедия характеров.
В комедии положений главным источником являются обстоятельства и события, в которые попадают лирические герои. В комедии характеров основой выступают характеры главных героев, зачастую их однобокость, недалекость, или любая другая гипертрофированная негативная черта.
Часто комедия характеров – сатирическая комедия, в которой тонко высмеиваются все негативные качества человека.
Виды юмора: ирония, сатира, сарказм
Понятие юмор следует рассматривать в широком и узком векторе. В широком смысле понятие юмор – это все, что вызывает смех или веселое расположение уха. В более узком смысле – это явление, которое подчеркивает комические стороны предметов, событий и личностей.
Одними из самых известных форм юмора, которые успешно применяются в драматургии, в частности, комедии – это ирония, сарказм и сатира. На первый взгляд эти понятия весьма похожи друг на друга своими функциями. Однако каждый из них представляет отдельный инструмент для создания комической ситуации.
Ирония – это вид юмора, который основан на противоречиях с действительным положением вещей, она вызывает насмешку над людьми, мнение которых остро противоречит действительности, подтверждая его. Например: «Да-да, ты очень сильный» — иронический прием, который высмеивает слабого человека, который считает себя сильным.
Сатира – вид юмора, который открыто, высмеивает ту или иную ситуацию, либо человека. Следует отметить, что сатира сама по себе является также разновидностью критики, однако, несколько смягченной по сравнению с ней.
Сарказм – это синтез иронии и сатиры, насмешка, имеющая негативную окраску. Сарказм не завуалировано подчеркивает негативные качества либо события. В отличие от иронии и сатиры, сарказм применяется в комедиях довольно редко, так как имеет откровенный негативный подтекст.
Виды юмора как способ создания характера героя
Главный герой комедии – человек, обладающий гипертрофированными низкими качествами, которые вызывают осуждение у других людей. Посредством применения в комедии юмора, в частности, сатиры, иронии и сарказма, автор словно подчеркивает всю низость главного героя, неверность его суждений, малообразованность.
Юмористические инструменты усиливают антагонистическое противостояние в комедии, делают его более ярко выраженным. Применяя сатиру и сарказм, автор выражает собственное мнение на счет нелепости ситуации или несовершенства главного героя, предоставляя, таким образом, читателю возможность сделать ранний вывод на счет развязки комедии.
Нужна помощь в учебе?
Предыдущая тема: Булгаков «Собачье сердце»: проблема нравственного сознания личности Следующая тема:   Поиск смысла жизни в литературе: идеал и действительность в литературе
Структура смеховой культуры Текст научной статьи по специальности «Языкознание и литературоведение»
УДК 008 РЕДКОЗУБОВА О.С. СТРУКТУРА СМЕХОВОЙ КУЛЬТУРЫ
Редкозубова Ольга Сергеевна, аспирантка Тамбовского государственного университета имени Г.Р. Державина
Аннотация. Статья посвящена структуре смеховой культуре. Смеховая культура, прежде всего -коллективное понятие; по своей природе она является ярко выраженным социокультурным явлением, выполняющим коммуникативную функцию. Через призму смеховой культуры человек получает ту или иную информацию. И как любое явление, она имеет свою структуру. В смеховую структуру входит юмор, ирония, остроумие, сатира, сарказм, пародия, карикатура и каламбур. Все данные формы неотъемлемая часть смеховой культуры российского студенчества. Многообразие оттенков смеха (юмор, сатира, ирония, сарказм, шутка, насмешка, гротеск каламбур) отражает богатство действительности. Формы и мера смеха определяются и объективными свойствами предмета, и мировоззренческими принципами человека, его отношением к миру, и национальными традициями культуры народа. Юмор, ирония, сатира, пародия, сарказм предстают как формы целостного смеха. Автор предлагает свою схему структуры смеховой культуры.
Смеховая культура XX века обладает рядом особенностей. Расширяется спектр осмеиваемых явлений в сравнении с предыдущими периодами: наряду с бульварным, поверхностным осмеянием всего непонятного возникают новые формы смеховой культуры, ориентированные на высокообразованную аудиторию, на взыскательного читателя и зрителя. Смеховая культура несет на себе отпечаток времени, когда обостряются противоречия между цивилизациями и идет формирование глобальной культуры.
Информационные технологии, приобщая человека к всемирному сообществу, снижают эмоциональную окраску восприятия, события (человек остается наедине с самим собой). Однако смеховая культура в силу своих особенностей может компенсировать негативные стороны информационного образа жизни, давая «живой» отклик на происходящее.
Общечеловеческие начала смеховой культуры помогают избежать ненужных разногласий, сгладить противостояние в мире. Смех является одним из начал, влияющих на целостность культурной системы.
Смех затрагивает все стороны человеческой жизни, мы смеемся, когда нам хорошо, когда не хотим показать собственный страх, боль, переживания и др. Особенно это проявляется в присутствии других людей, при которых мы стараемся «сохранить» лицо — как говорят японцы. Существуют физиологические, психологические аспекты смеха. С помощью смеха мы освобождаемся от страха, волнения. И в тоже время смех имеет большую социальную значимость. Смех является категорией понимания. Например, юмор студентов может понять лишь студент, смеховая культура того или иного общества несет свои специфические черты. Одновременно с этим, смех можно рассматривать как понимание того, каким образом можно решить проблему, как выйти из сложной и запутанной ситуации.
Смеховая культура, прежде всего — коллективное понятие; по своей природе она является ярко выраженным социокультурным явлением, выполняющим коммуникативную функцию. Через призму смеховой культуры человек получает ту или иную информацию.
И как любое явление, она имеет свою структуру.
Достаточно часто в теории смеха используется понятие «юмор». Именно юмор занимает важное место в смеховой структуре. В современных исследованиях понятие «юмор» имеет два значения. В узком смысле юмор обозначает один из видов комического, который, как правило, характеризуется сочувственным отношением к объекту насмешки. В широком значении под юмором понимают способность человека или социальной группы воспринимать комическое во всем его разнообразии.
В целом юмор стремится к сложной, как сама жизнь оценке, свободной от односторонностей общепринятых стереотипов. На более глубоком (серьёзном) уровне юмор открывает за ничтожным возвышенное, за безумным мудрость, за своенравным подлинную природу вещей, за смешным грустное — «сквозь видный миру смех… незримые ему слезы» (по словам Н. В. Гоголя). Жан Поль, первый теоретик юмора, уподобляет его птице, которая летит к небу вверх хвостом, никогда не теряя из виду землю, — образ, материализующий оба аспекта юмора. Юмор — признак самой жизни в ее развитии, движении, переломах и конфликтах, противостоящий всякой статике и теоретизированию.
В зависимости от эмоционального тона и культурного уровня юмор может быть добродушным, жестоким, дружеским, грубым, печальным, трогательным и т. п. «Текучая» природа юмора обнаруживает «протеическую» (Жан Поль) способность принимать любые формы, отвечающие умонастроению любой эпохи, её историческому «нраву», и выражается также в способности сочетаться с любыми иными видами смеха: переходные разновидности юмора иронического, остроумного, сатирического, забавного. Сопоставление с основными видами комического многое уясняет в существе и своеобразии чистого юмора. Чувство юмора как разновидность эстетического чувства всегда опирается на высокие эстетические идеалы. В противном случае юмор превращается в скепсис, цинизм, сальность, пошлость, скабрезность. Юмор предполагает способность хотя бы эмоционально, в самой общей эстетической форме схватывать противоречия действительности. Юмор присущ эстетически развитому уму, способному быстро, эмоционально -критически оценивать сущность явления, склонному к богатым и неожиданным сопоставлениям и ассоциациям.
Активная, творческая форма чувства юмора — остроумие. Если юмор — это способность к восприятию комизма, то остроумие — к его творению, созиданию. Остроумие — это талант так концентрировать, заострять и эстетически оценивать реальные противоречия действительности, чтобы нагляден, ощутим стал их комизм. Например, в студенческой среде всегда популярны, так называемые «душа компании», «шуты», которые всегда подшучивают над однокурсниками и преподавателями. У таких студентов не бывает проблем с учебой и в отношениях на курсе, они легко выходят из нелепых ситуаций, потому что в любой проблеме видят комическое и смешное, а самое главное помогают увидеть это другим.
Следующий элемент структуры смеха — это ирония. Ирония является выражением объективного контраста, скрывающего за собой субъективность. В иронии смешное скрывается под маской серьёзности — с преобладанием отрицательного (насмешливого) отношения к предмету; в юмор серьёзное — под маской смешного, обычно с преобладанием положительного («смеющегося») отношения. Сложность иронии лишь формальная, её серьёзность — мнимая, её природа — чисто артистическая; напротив, сложность юмора содержательная, его серьёзность — подлинная, его природа — даже в игре — скорее «философическая», мировоззренческая. Юмор нередко «играет» на двух равно действительных аспектах человеческой натуры — физическом и духовном. Различен поэтому эффект иронии и юмора, когда игра закончена и обнажается внутренний аспект, подлинная цель игры: ирония, порой близкая смеху язвительному, задевает, ранит, оскорбляет сугубо — не только открывшимся неприглядным содержанием, но и самой формой игры: тогда как юмор в конечном счёте заступается за свой предмет, а его смехом иногда, например в «дружеском» юмор, «стыдливо» прикрывается восхищение, даже прославление. Колоритом юмора художники нового времени часто пользуются — во избежание ходульности или односторонности — для изображения наиболее благородных героев, а также идеальных натур «простых людей», национально или социально характерных.
Высшая степень иронии, открывающая негативную оценку явления непосредственно вслед за позитивным «зачином» (который ирония в остальных случаях сохраняет и развивает, припрятывая критицизм в подтекст, в подразумеваемое) — это сарказм. По тенденциозности сарказм всегда беспощаден, сатиричен, тогда как ирония при использовании её в интересах юмора усваивает мягкие, умиротворённые тона.
Достаточно близко к сфере смешного стоит сатира. Сатира появляется при условии перехода из царства рассудка в сферу морали. Здесь «бесконечно-малое», ничтожное, лежащее в основе смеха является не заблуждением, а пороком или злом и вызывает уже не удовольствие, а негодование. Смех перестает быть смехом — он трансформируется в осмеяние и моральное неодобрение. Сатира (лат. satira, от более раннего satura — сатура, буквально — смесь, всякая всячина), вид комического; беспощадное, уничтожающее переосмысление объекта изображения (и критики), разрешающееся
смехом, откровенным или подспудным, «редуцированным»; специфический способ художественного воспроизведения действительности, раскрывающий её как нечто превратное, несообразное, внутренне несостоятельное (содержательный аспект) посредством смеховых, обличительно-осмеивающих образов (формальный аспект).
В отличие от прямого обличения, художественная сатира как бы двусюжетна: комическое развитие событий на первом плане предопределяется некими драматическими или трагическими коллизиями в «подтексте», в сфере подразумеваемого. Двусюжетны и другие виды комического, используемые в сатирическом произведении: юмор и ирония; но для собственно сатиры характерна негативная окрашенность обоих сюжетов — видимого и скрытого, тогда как юмор воспринимает их в позитивных тонах, ирония — комбинация внешнего позитивного сюжета с внутренним негативным.
Сатира — насущное средство общественной борьбы; актуальное восприятие. Сатира в этом качестве — переменная величина, зависимая от исторических, национальных и социальных обстоятельств. Но чем всенароднее и универсальнее идеал, во имя которого сатирик творит отрицающий смех, тем «живучей» сатира, тем выше её способность к возрождению. Эстетическая «сверхзадача» сатира — возбуждать и оживлять воспоминание о прекрасном (добре, истине, красоте), оскорбляемом низостью, глупостью, пороком. Духовно «пристыжая» и очищая смеющегося, сатира тем самым защищает положительное, подлинно живое. По классическому определению Ф. Шиллера, впервые рассмотревшего сатиру как эстетическую категорию, «… в сатире действительность как некое несовершенство противополагается идеалу, как высшей реальности». Но идеал сатирика выражен через «антиидеал», т. е. через вопиюще-смехотворное отсутствие его в предмете обличения.
Бескомпромиссность суждений о предмете осмеяния, откровенная тенденциозность -присущий именно сатире способ выражения авторской индивидуальности, стремящейся установить непереходимую границу между собственным миром и предметом обличения, и «… силой субъективных выдумок, молниеносных мыслей, поразительных способов трактовки разложить все то, что хочет сделаться объективным и приобрести прочный образ действительности…» (Гегель, Эстетика, т. 2, М., 1969, с. 312).
Отчётливо осознавшая себя в древнеримской литературе в качестве обличительно-насмешливого жанра лирики, позже сатира, сохраняя черты лиризма, утратила жанровую определённость, превратилась в подобие литературного рода, определяющего специфику многих жанров: басни, эпиграммы, бурлески, памфлета, фельетона, сатирического романа.
Сатирик «моделирует» свой объект, создавая образ, наделённый высокой степенью условности, достигаемой «направленным искажением» реальных контуров явления с помощью преувеличения, заострения, гиперболизации, гротеска. Сатира очень популярна в студенческой среде. Студенты, очень часто используют данный вид смеховой культуры на своих вечеринках, КВНах, капустниках, тем самым притягивая общественность к интересующим их вопросам. Очень часто в шуточной форме затрагиваются вопросы о размерах стипендии, сдаче сессии и многое другое. Отношение юмора к сатире определяется уже тем, что источником сатирического смеха служат пороки, недостатки как таковые, а юмор исходит из той истины, что наши недостатки и слабости — это чаще всего продолжение, утрировка или изнанка наших же личных достоинств. Сатира, открыто разоблачая объект, откровенна в своих целях, тенденциозна, тогда как серьёзная цель юмора, глубже залегая в структуре образа, более или менее скрыта за смеховым аспектом. a, буквально — пение наизнанку), в литературе и (реже) в музыкальном и изобразительном искусстве комическое подражание художественному произведению или группе произведений. Обычно пародия строится на нарочитом несоответствии стилистических и тематических планов художественной формы; два классических типа пародии (иногда выделяемые в особые жанры) — бурлеска, низкий предмет, излагаемый высоким стилем и травестия, высокий предмет, излагаемый низким стилем Осмеяние может сосредоточиться как на стиле, так и на тематике — высмеиваются как заштампованные, отставшие от жизни приёмы поэзии, так и пошлые, недостойные поэзии явления действительности; разделить то и другое иногда очень трудно. Пародироваться может поэтика конкретного произведения автора, жанра, целого литературного
направления, целого идейного миросозерцания. По характеру комизма пародия может быть юмористической и сатирической, со многими переходными ступенями. Пародия помогает трансформировать в человеке, предмете, явлении недостатки в положительное. Студенты, часто обращаются к пародии. Предметом пародии являются преподаватели, администрация учебного заведения. Зачастую с в пародии студенты видят некое выражении свободы, пародия помогает встать отношениям студентов и преподавателей на другой уровень.
Популярна в молодежной среде — карикатура (итал. сапсаШга, от сапсаге — нагружать, преувеличивать), способ художественной типизации, использование средств шаржа и гротеска для критически целенаправленного, тенденциозного преувеличения и подчёркивания негативных сторон жизненных явлений или лиц; в карикатуре, составляющей специфическую область проявления комического в изобразительном искусстве, сатира и юмор служат для критики, разоблачения, осмеяния каких-либо социальных, общественно-политических, бытовых явлений. Данная форма смеховой культуры крепко закрепилась в России, особенно в студенческой среде. Студенты всегда рисовали преподавателей и своих друзей. Карикатура в студенческой среде всегда носила позитивный характер. В широком смысле слова под карикатурой понимают всякое изображение, где сознательно создаётся комический эффект, соединяются реальное и фантастическое, преувеличиваются и заостряются характерные черты фигуры, лица, костюма, манеры поведения людей, изменяются соотношения их с окружающей средой, используются неожиданные сопоставления и уподобления. Карикатура в этом значении обладает широчайшим диапазоном тем и может быть сопоставлена с карнавальным действом, театральной буффонадой, литературным бурлеском и эпиграммой.
Многие исследователи комического (З. Фрейд, К. Фишер, Т. Липпс) относят каламбур к низшему сорту шутки. Однако для Франции 17 — 18 вв. каламбур был высшей формой остроумия. Его легкость, блеск, беззаботная веселость эстетически соответствовали характеру жизни высших слоев общества, определявших духовную жизнь нации. Способность каламбурить высоко ценилась и служила своеобразной визитной карточкой человека. Существует притча: Людовик IV захотел испытать остроумие одного придворного и сказал ему, что он, король, хочет быть сам сюжетом остроты. В ответ кавалер удачно скаламбурил: «Король — не сюжет, король — не подданный». Во французском языке слово <«и)еЪ> одновременно означает «сюжет» и «подданный». Отсюда игра слов в ответе. Это характерный пример французского галантного остроумия.
В конце 18 в. Великая французская революция вместе с королевским двором смела и галантный аристократический каламбур. Господствовать в области комизма стал гротеск. Его острие зло жалило аристократию. Святыни монархической государственности были повержены, освистаны и осмеяны с высоты идеалов всеобщей свободы, равенства и братства. Однако в середине 19 в. стало ясно, что эти идеалы не осуществились, хотя и ценности аристократического прошлого померкли безвозвратно. Безверие и отсутствие ясных идеалов породили во Франции особый род остроумия -благг. Эта беспощадная насмешка над тем, чему люди привыкли поклоняться, — дитя общественных разочарований. Утраченные иллюзии стали обыкновенной историей, а в сфере юмора это выразилось в безрадостном, подернутом цинизмом смехе, для которого нет ничего заповедного, неприкосновенного. Характерный пример благга: «Эта женщина как республика, она была прекрасна во времена империи».
В 20 в. возник гегг — новая форма юмора, окрашенного неопасным ужасом, отражающего отчуждение людей в индустриальном обществе. Вот типичный американский рекламный рассказ, построенный по принципу гегга. Два враждующих машиниста повели навстречу друг другу поезда, полные пассажиров. На полотно выбегает ребенок с мячом. Поезда сталкиваются, но… катастрофы не происходит, они разлетаются в разные стороны благодаря мячу. «Покупайте мячи фирмы такой — то!» На этом же принципе построены знаменитые кадры путешествия Чарли Чаплина между шестернями огромной машины в кинофильме «Новые времена». Под влиянием американской культуры гегг получил распространение и в смеховой культуре Франции. Данный вид смеховой культуры получил в современной российской студенческой среде большую популярность. Студенты уже сегодня используют его в своих вечерах.
Каламбур, гротеск, благг, гегг — формы французского юмора, обусловленные характером жизни нации на разных этапах ее развития. Сказанное, конечно, не значит, что гротеск не существовал раньше или что каламбур умер вместе с падением аристократии. Нет, речь идет лишь о преимущественном развитии тех или других оттенков комизма, той или иной эстетики остроумия в разные периоды развития страны.
Особенности национального своеобразия культуры каждого народа заключаются не столько в его одежде или кухне, сколько в манере понимать вещи. Эта манера понимать вещи отчетливо и выпукло проявляется в национально окрашенных формах комизма.
Комическое национально своеобразно, и в то же время в нем проступают интернациональные и общечеловеческие черты. В силу общности законов социального развития часто одни и те же явления с одинаковой непримиримостью высмеивают все народы.
Многообразие оттенков смеха (юмор, сатира, ирония, сарказм, шутка, насмешка, каламбур) отражает богатство действительности. Формы и мера смеха определяются и объективными свойствами предмета, и мировоззренческими принципами человека, его отношением к миру, и национальными традициями культуры народа.
Юмор, ирония, сатира, пародия, сарказм предстают как формы целостного смеха.
Взаимодействие человека с реальным миром сложно и многообразно. Обстоятельства текучи и изменчивы, а человек, оставаясь самим собой, в каждой ситуации равен и не равен себе, он тот же и вместе с тем иной. Все элементы смеховой культуры связаны с различием человеческих отношений, а они составляют движущую силу культуры.
Структура смеховой культуры едина для всех культурных эпох, каким бы различным ни было их наполнение. Творческая, жизнетворящая сила смеха давно подмечена людьми. Через все формы смеха мы общаемся, обмениваемся информацией, приобретаем новые знакомства. А в «Подростке» Ф.М.Достоевского мы и вовсе обнаруживаем готовую формулу: «…Если захотите рассмотреть человека и узнать его душу, то вникайте не в то, как он молчит, или как он говорит, или как он плачет, или даже как он волнуется благороднейшими идеями, а высмотрите лучше его, когда он смеется. Хорошо смеется человек — значит хороший человек. … Смех есть самая верная проба души».
СТРУКТУРА СМЕХОВОЙ КУЛЬТУРЫ.
Выставка «Сатира, юмор и ирония Лескова»
Регион:
Воронежская область
Источник:
ГБУК ВО «Воронежская областная юношеская библиотека имени В. М. Кубанева»
Адрес:
Воронеж, ул Никитинская, д 32
Стоимость:
Бесплатно ₽
Категория:
Выставки
Время проведения:
События завершились
Место проведения:
Воронежская областная юношеская библиотека имени В. М. Кубанёва
Возрастное ограничение:
12+
Автор фотографии: Ольга Романцова;Источник: Архив Воронежской областной юношеской библиотеки им. В. М. Кубанева
Н. С. Лесков – самобытный писатель, создатель целой галереи образов русских «праведников», среди которых самый яркий – простой тульский мастер Левша, сумевший подковать стальную блоху.
На выставке читатели найдут произведения великого рассказчика, включенные в состав 12-томного собрания сочинений. Среди них – «Очарованный странник» – повесть, в которой автор создает особый образ человека, несопоставимого ни с одним из героев русской литературы.
Особого внимания заслуживает книга, созданная сыном писателя, в которой содержатся ценнейшие биографические сведения о Н. С. Лескове.
Выставка предназначена для широкого круга читателей.
Валерий Казаков — Уроки жизни. Юмор, сатира, ирония читать онлайн бесплатно
Создано в интеллектуальной издательской системе Ridero
Откровенно говоря, мне никогда не нравились успешные люди. Я всегда замечал в них нечто ущербное. Некую отстраненность от реальной жизни. Скорее всего это происходило потому, что сам я никогда не обладал статусом делового человека. Я всегда был катастрофически неуспешен и потому вынужден был находиться в народной гуще.
Я никогда не имел престижной должности. За всю свою жизнь я ни разу не пришел на службу в новом костюме и не примерил перед зеркалом красивый новый галстук. Сколько себя помню, на мох плечах всегда был ватник, на ногах – кирзовые сапоги. В этом смысле жизнь на севере России не отличается большим разнообразием в выборе одежды. Просто летом мне нужна фуфайка потоньше и сапоги полегче. Вот и всё.
Встав с постели, я никогда не говорил жене: «С добрым утром». Она мне тоже ничего не говорила. Молча, ставила на стол тарелку овсяной каши, нарезала хлеб, я тянулся за маслом. Мы ели и смотрели в разные стороны. Потом мы пили чай. Она говорила, что ей пора на работу. Я кивал. Она уходила. Я не провожал её даже глазами.
За всю свою жизнь я никогда не приходил на работу к восьми часам. Моя работа всегда была трудной, но не имела строгих временных рамок.
В бригаде лесорубов я много выпивал. В школьной котельной дрался с пьяными бомжами, которые лезли в тепло отогреться. Работая лесником, я старался исполнить свои обязанности поскорее, чтобы к обеду выбраться из чащи, где полно комаров и мошек.
Сколько себя помню, я некогда не имел больших денег, хотя порой хорошо зарабатывал. Мои деньги как-то очень быстро уходили на самые неотложные нужды. Что называется – исчезали. И когда мне становилось необходимо купить бритву или вылечить зуб, то в семейном бюджете денег на эти цели часто не находилось. Появлялись они только после моих грубых выкриков и громкой брани. Потому что я, оказывается, отнимал у жены последнее. Потому что я, по её мнению, всегда был транжира и мот, который принесет денег с Гулькин нос и тут же их забирает обратно.
Я никогда не бил жену, даже если она очень меня раздражала. Я просто надевал фуфайку, кирзовые сапоги и выходил на улицу в сад. Там я долго стоял под раскидистой яблоней, опираясь на черен лопаты. Я ни о чем не думал, ни о чем не жалел. Просто так складывалась моя жизнь.
Я никогда не говорил жене ласковых слов. Мне как-то неудобно было такие слова говорить. Потому что жена через два годы после нашей свадьбы стала толстой, крикливой, вечно чем-то недовольной женщиной. Иногда спросонья я смотрел на неё и не узнавал. Мне казалось, что её подменили. Я женился на тонкой изящной, милой женщине, от которой пахло ландышами, которая с придыханием говорила о Чайковском и Рахманинове, восторгалась Джойсом и Набоковым, которая рассказывала о последнем лауреате Пулитцеровской премии по литературе – Соле Беллоу. А сейчас она не может вспомнить, кто такой Мендельсон, не говоря уже о Рее Брэдбери. Она говорит со мной о своих больных коленях, о высоком давлении, о нехватке денежных средств. От неё пахнет ужасной мазью под названием «Диклоран плюс». И я боюсь спросить у неё, любит ли она меня, как прежде, или это чувство уже совершенно угасло в ней, погребенное под грудой неотложных дел и забот.
Среди моих одноклассников очень мало успешных людей. Может быть, когда-то они и были успешными, но сейчас все мои друзья и бывшие одноклассники почему-то стали пенсионерами. Глядя на них, я со страхом думаю: «Неужели я выгляжу так же непрезентабельно, как эти люди»? И порой мне кажется, что это не так. Что я выгляжу лучше и моложе. Что они всегда выглядели хуже меня. Их мучила изжога, они много болели, злились, переживали из-за пустяков, стремились достичь успеха. А мне все эти устремления были чужды.
И тут вдруг я понимаю, что давно не видел себя со стороны.
Я спешу к зеркалу и придирчиво разглядываю в нем свое лицо. Седеющие волосы, тонкие скулы, глубоко посаженные темные глаза. Скорее всего кто-то из моих предков был турком или иранцем. Не иначе. Во всяком случае, у меня не растет окладистая борода, как у всех породистых славян. И вообще мне порой хочется взять саблю, вскочить на резвого коня и скакать, скакать по широкой степи навстречу ветру. «Эх, ма! Степь раздольная»! Но коня у меня нет. Нет даже сабли. Зато в душе есть что-то такое, отчего я не могу смотреть на породистых коней без восхищения. Мне нравятся их вытянутые интеллигентные морды, их большие, влажные добрые глаза. Их мягкие теплые губы, длинные ресницы. Мне нравится слушать, как они ржут. Видеть, как они едят овес, пьют воду из реки. Должно быть, это проявляют себя мои турецкие корни.
И живу я почему-то не в Париже, не в Москве и даже не в Малмыже. Я живу в селе под странным названием Трек. Скорее всего, это тоже неспроста. Ведь по логике вещей турки должны жить именно в Туреке.
Может быть кому-то мое предположение о турецких корнях покажется неубедительным, даже странным. Спорить не буду, некоторые мои умозрительные заключения носят чисто экзистенциальный характер. Но почему-то они мне дороги. Они не противоречат моему внутреннему «я». Поэтому я вам как турок скажу. Русский народ живет неправильно. Он излишне патриотичен. Для него имперская Россия превыше всего.
Русский народ отвоевал когда-то у нас, у турков, Крым. И что из этого получилось? Ничего хорошего. Дальше последовали только новые войны, которые не кончаются до сих пор. Россия всегда в этом смысле выполняла не свою роль. Воплощала не свои планы. Создавала не свой образ. То есть её амбиции не соответствовали её мощи. При нищем народе она вооружала свою армию так, что та могла держать в страхе всю Европу. Только эта ноша всегда была ей не по силам, не по плечу. И тогда, и сейчас.
Вот пишу эти строки, а сам понимаю, что я никогда не имел одного устоявшегося мнения на что-либо. Сегодня я думаю так, завтра – иначе, а через неделю, вполне возможно, заброшу все эти мысли к чертовой бабушке. Я человек непостоянный. Я свободен от каких бы то ни было принципов. А полная свобода, на мой взгляд, как раз и предполагает идейное непостоянство. Сегодня я могу думать так, завтра иначе, а послезавтра вообще могу забыть обо всем, что тревожило меня ранее. Потому что от дум моих ничего не меняется. Думы мои легки и оттого горьки. Из под моей руки – буковки, завитки.
Я никогда не рассуждал о жизни серьёзно. Серьёзная жизнь не для меня. Когда я пробую рассуждать о своей жизни всерьёз – мне всегда хочется плакать. Ибо суверенность моего существования достигается как раз за счет отсутствия ясной перспективы. Четкие очертания будущего навевают на меня тоску. У какого-то писателя я выудил фразу о том, что достижимые вещи обычно губят самое богатое воображение. На первый взгляд эта мысль может показаться нелепой. Но это не так. Подумайте сами, что раньше времени приземляет нашу высоко парящую душу? Забота о деньгах. А что дают нам деньги? Еду. Всего лишь еду. А мы, порой, тратим на это целую жизнь. Значит, мы свою жизнь проедаем. Мы проедаем вечность, славу, признание, мечту.
это… Примеры иронии в литературе
Ирония является одним из видов тропов. Ирония — это художественный прием создания образной и выразительной речи на основе отождествления предметов по контрасту, а не по сходству признаков, как в метафоре, или по смежности, как в метонимии.
Греческое слово eironeia буквально значит «притворство». Как же создается ирония в языке и художественной речи?
Этот троп встречается гораздо реже, чем метафоры или метонимии. Говорящий или художник слова с помощью иронии высмеивают какое-то явление или событие, при этом называя его не тем словом, которого оно заслуживает, а совсем наоборот, глупец называется умным, что-то мелкое и не заслуживающее внимания назовется большим.
Ирония — это скрытая насмешка
Определение
Ирония — это употребление слова или оборота речи в противоположном значении.
Происходит нарочитое «переименование», которое выражает насмешливое или вовсе негативное отношение говорящего к обсуждаемому предмету, например:
зайдите в мои хоромы (приглашение зайти в небольшую квартиру);
вот идет большой человек (о младенце, только научившемся ходить);
люблю как собака палку;
всю жизнь об этом мечтал!
только об этом и думаю!
кому нужна такая красота.
Ирония, как троп, часто используется в художественной литературе в виде слов и словосочетаний с положительным значением для отрицательной характеристики человека, для создания сатирического образа. Можно утверждать, что ирония — это скрытое осуждение под видом похвалы. Она чаще всего выявляется в контексте и характеризуется яркой эмоциональной окраской.
Видеоурок «Отличие иронии от сарказма»
Ирония в художественной литературе. Примеры
Классик русской литературы Н.В. Гоголь в поэме «Мёртвые души» с совершенно с серьёзным видом повествует о полицмейстере-взяточнике:
Полицмейстер был некоторым образом отец и благотворитель в городе. Он был среди граждан совершенно как родной в семье, а в лавки и в гостиный двор наведывался, как в собственную кладовую.
Тонкая, замаскированная за внешней учтивостью насмешка проглядывает в ироничном описании А.С. Пушкиным высшего света Петербурга:
Тут был, однако, цвет столицы, И знать, и моды образцы, Везде встречаемые лицы, Необходимые глупцы.
Или вот еще яркий пример пушкинской иронии:
Старик, имея много дел, В иные книги не глядел.
А далее выясняется, какие же это «дела»:
Он в том покое поселился, Где деревенский старожил Лет сорок с ключницей бранился, В окно смотрел да мух давил.
Знаменитый баснописец И.А. Крылов иронично утверждает противоположное тому, что думают об его персонаже:
— Отколе, умная, бредешь ты, голова? — Лисица, встретяся с Ослом, его спросила.
В известной басне звучит тонкая затаенная насмешка автора.
— Ты всё пела? Это — дело, —
иронически говорит Муравей Стрекозе, считая в действительности пение бездельем.
Видеоурок «Юмор, сатира, ирония, сарказм»
Скачать статью: PDF
основной конфликт в литературных произведениях с примерами и какие его отличия или что общего у героев
Комедия представляет собой вид драмы, который отличается сатирическим и юмористическим подтекстом, через который происходит развязка борьбы антагонистических персонажей либо же действенного конфликта.
Комедия, как драматургический жанр, возникла еще в Древней Греции. Аристотель определял термин «комедия» как демонстрация людям их же пороков в юмористическом виде. В драматургии различают два вида комедий: комедия положений и комедия характеров.
Юмор как способ создания характера героя
В комедии положений главным источником являются обстоятельства и события, в которые попадают лирические герои. В комедии характеров основой выступают характеры главных героев, зачастую их однобокость, недалекость, или любая другая гипертрофированная негативная черта.
Часто комедия характеров – сатирическая комедия, в которой тонко высмеиваются все негативные качества человека.
Виды юмора: ирония, сатира, сарказм
Понятие юмор следует рассматривать в широком и узком векторе. В широком смысле понятие юмор – это все, что вызывает смех или веселое расположение уха. В более узком смысле – это явление, которое подчеркивает комические стороны предметов, событий и личностей.
Одними из самых известных форм юмора, которые успешно применяются в драматургии, в частности, комедии – это ирония, сарказм и сатира. На первый взгляд эти понятия весьма похожи друг на друга своими функциями. Однако каждый из них представляет отдельный инструмент для создания комической ситуации.
Ирония – это вид юмора, который основан на противоречиях с действительным положением вещей, она вызывает насмешку над людьми, мнение которых остро противоречит действительности, подтверждая его. Например: «Да-да, ты очень сильный» — иронический прием, который высмеивает слабого человека, который считает себя сильным.
Сатира – вид юмора, который открыто, высмеивает ту или иную ситуацию, либо человека. Следует отметить, что сатира сама по себе является также разновидностью критики, однако, несколько смягченной по сравнению с ней.
Сарказм – это синтез иронии и сатиры, насмешка, имеющая негативную окраску. Сарказм не завуалировано подчеркивает негативные качества либо события. В отличие от иронии и сатиры, сарказм применяется в комедиях довольно редко, так как имеет откровенный негативный подтекст.
Виды юмора как способ создания характера героя
Главный герой комедии – человек, обладающий гипертрофированными низкими качествами, которые вызывают осуждение у других людей. Посредством применения в комедии юмора, в частности, сатиры, иронии и сарказма, автор словно подчеркивает всю низость главного героя, неверность его суждений, малообразованность.
Юмористические инструменты усиливают антагонистическое противостояние в комедии, делают его более ярко выраженным. Применяя сатиру и сарказм, автор выражает собственное мнение на счет нелепости ситуации или несовершенства главного героя, предоставляя, таким образом, читателю возможность сделать ранний вывод на счет развязки комедии.
Трагическое и комическое. Сатира, юмор, ирония, сарказм. Гротеск
Трагическое и комическое
Трагическое – 1. Эстетическая категория, в основе которой – наличие противоречия, конфликта в жизни человека или его отношениях, который не может быть разрешен, но с которым нельзя примириться.
Последствия этого конфликта могут быть катастрофичны для героя, но его несгибаемое мужество перед лицом неминуемого поражения становится основанием для веры и надежды зрителей или читателей. 2.
Все ужасное в человеческой жизни, вызывающее боль, страдание, отмеченное острыми столкновениями человека с миром; катастрофичность человеческого бытия.
Комическое – эстетическая категория, отражающая противоречия, присущие явлениям действительности, и выражающая их отрицательную оценку; несоответствие реального и воображаемого, истинного и мнимого, внутреннего и внешнего; форма критики несовершенных явлений действительности. В основе комического – смех. По эмоциональным оттенкам делится на юмор, иронию, сатиру, сарказм.
Юмор – это вид комического, в котором пороки осмеиваются не беспощадно, как в сатире, а доброжелательно подчеркиваются недостатки и слабости человека или явления, напоминая о том, что они часто лишь продолжение или изнанка наших достоинств.
Ирония – это троп, состоящий в употреблении слова или выражения в смысле обратном буквальному с целью насмешки.
Отколе, умная, бредешь ты, голова! (Крылов) (в обращении к ослу).
Сатира – это разновидность литературы, специфическими формами обличающая и высмеивающая пороки людей и общества. Формы эти могут быть самыми разнообразными – парадокс и гипербола, гротеск и пародия и т.д.
Сарказм – это едкая язвительная насмешка над кем-либо или над чем-либо. Широко используется в сатирических литературных произведениях.
Гротеск – это причудливое смешение в образе реального и фантастического, прекрасного и безобразного, трагического и комического – для более впечатляющего выражения творческого замысла. Гротеск — изображение людей, предметов, деталей в литературе в фантастически преувеличенном, уродливо-комическом виде.
Гротеск обязательно нарушает границы правдоподобия, придает изображению некую условность и выводит художественный образ за пределы вероятного, осознанно деформируя его. Примеры – книги Ф. Рабле и Дж. Свифта.
В русской литературе широко пользовались гротеском при создании ярких и необычных художественных образов Н.В. Гоголь (Нос, Записки сумасшедшего), М.Е. Салтыков-Щедрин (История одного города, Дикий помещик и другие сказки), Ф.М. Достоевский (Двойник. Приключения господина Голядкина), Ф. Сологуб (Мелкий бес), М.А. Булгаков (Роковые яйца, Собачье сердце), А. Белый (Петербург, Маски), В.В. Маяковский (Мистерия-буфф, Клоп, Баня, Прозаседавшиеся), А.Т. Твардовский (Теркин на том свете), А.А. Вознесенский (Оза), Е.Л. Шварц (Дракон, Голый король).
Наряду с сатирическим, гротеск может быть юмористическим, когда с помощью фантастического начала и в фантастических формах внешности и поведения персонажей воплощаются качества, вызывающие ироническое отношение читателя, а также – трагическим (в произведениях трагического содержания, рассказывающих о попытках и судьбе духовного определения личности.
Трагическое и комическое. Сатира, юмор, ирония, сарказм. Гротеск
Трагическое – эстетическая категория, для которой характерно наличие неразрешимого конфликта. В центре внимания, как правило, страдания и гибель героя или его жизненных ценностей.
В отличие от печального или ужасного, трагическое вызывается не случайными внешними силами, а проистекает из внутренней природы человека.
Трагическое предполагает свободное действие человека, в результате которого он обрекает себя на страдания или смерть.
Казалось бы, суть трагедии Гамлета (трагедия Шекспира “Гамлет”) – в тех событиях, которые с ним произошли. Но подобные несчастья обрушились и на Лаэрта. Однако при этом нельзя говорить о том, что Лаэрт – трагический герой, поскольку он пассивен, а Гамлет сам, сознательно идет навстречу трагическим обстоятельствам.
Он выбирает схватку с “морем бед”. Именно об этом выборе и идет речь в знаменитом монологе:
Быть или не быть, вот в чем вопрос.
Достойно ль
Смиряться под ударами судьбы,
Иль надо оказать сопротивленье
И в смертной схватке с целым морем бед
Покончить с ними? Умереть. Забыться.
Комическое – средство раскрытия жизненного противоречия путем осмеяния. Основные виды комического: юмор, ирония, сатира, сарказм.
В основе комического всегда лежит какое-то несоответствие, нарушение нормы.
Это несоответствие может быть на языковом уровне (нелепицы, оговорки, имитация дефекта речи, акцента, не к месту звучащая иностранная речь), на уровне сюжетной ситуации (недоразумение, одного героя принимают за другого, неузнавание, ошибочные действия), на уровне характера (противоречие между самооценкой и произведенным впечатлением, между словом и делом, между желаемым и действительным и т. д.). Так, главный герой комедии Грибоедова “Горе от ума” Чацкий часто попадает в комические ситуации.
Его обличительные речи не всегда уместны. Впервые увидев Софью после долгой разлуки, Чацкий, влюбленный в нее, почему-то начинает разговор с нападок на ее родственников и т. д.
Юмор – особый вид комического, изображение героев в смешном виде. В отличие от сатиры, юмор – смех веселый, добродушный, помогающий человеку освободиться от предрассудков, ошибочных убеждений, недостатков. Так, гоголевская повесть “Ночь перед Рождеством” буквально пронизана юмором (описание капризной красавицы Оксаны, Чуба и т. д.).
Ирония – особый вид комического, осмеяние, насмешка. При иронии отрицательный смысл скрыт за внешней положительной формой высказывания. Например, в “Мертвых душах” Гоголь иронически изображает помещиков и чиновников.
Ирония в характеристике Ноздрева заключается в противоречии между ее первой частью, где подобные Ноздреву люди называются хорошими товарищами, и последующими словами о том, что они “при всем том бывают весьма больно поколачиваемы”.
Сатира – особый вид комического: высмеивание, разоблачение отрицательных сторон жизни, изображение их в нелепом, карикатурном виде. Например, явно сатирично описан в “Мастере и Маргарите” Булгакова “дом Грибоедова”, где размещалась ассоциация писателей МАССОЛИТ. О литературе здесь мало что напоминает, а все двери увешаны табличками типа “Рыбно-дачная секция”.
Сарказм – особый вид комического, язвительная насмешка, высшая степень иронии, когда негодование высказывается вцолне открыто.
Например, с сарказмом говорит Лермонтов в стихотворении “Дума” о своем поколении: “Богаты мы, едва из колыбели, Ошибками отцов и поздним их умом…”, и заканчивет едким сравнением отношения к нему будущих поколений с “Насмешкой горькою обманутого сына Над промотавшимся отцом”.
Гротеск – литературный прием, соединение реального и фантастического, создающее абсурдные ситуации, комические несоответствия. Например, гротеск активно используется Салтыковым-Щедриным в “Истории одного города”.
По сути, все события, происходящие в городе, являются гротеском.
Жители города, глуповцы, “чувствуют себя сиротами” без градоначальников и считают “спасительной строгостью” бесчинства Органчика, который знает только два слова – “не потерплю” и “разорю”.
Вполне приемлемыми кажутся горожанам такие градоначальники, как Прыщ с фаршированной головой или француз Дю-Марио. Однако своей кульминации абсурдность достигает при появлении Угрюм-Бурчеева, задумавшего захватить всю вселенную.
Стремясь реализовать свой “систематический бред”, Угрюм-Бурчеев пытается все в природе уравнять, так устроить общество, чтобы все в Глупове жили по придуманному им самим плану, что в результате приводит к разрушению города его же жителями.
Трагическое и комическое. Сатира, юмор, ирония, сарказм. Гротеск
Комическое в произведении искусства выражается в осмеянии каких-либо явлений, которые автор считает порочными.
Смех вызывают отдельные персонажи, ситуации, фрагменты действия. Подобный прием – сатирическое описание, сатира.
Эффект от уничтожающей силы сатиры получается тем больше, чем успешнее автор использует выразительные языковые средства: гротеск, гиперболу, пародию, сарказм. Классической сатирической литературой считаются произведения М.Е. Салтыков – Щедрина, некоторые рассказы А.П. Чехова, М.Зощенко и других мастеров слова.
Юмор также является способом проявления смешного в искусстве, однако юмористическое произведение несколько отличается от сатирического: если сатира осмеивает, уничтожает смехом какой- либо порок, то юмор лишь указывает на недостаток, посмеиваясь над ним добродушной усмешкой.
Ирония разоблачает отрицательные стороны чего – либо, делает это либо с горьким чувством, либо с доброй усмешкой.
Ирония помогает автору за внешней положительностью персонажа или явления скрыть их подлинное «лицо» (таковы помещики в Мертвых душах»).
Сарказм – особый вид проявления комического начала, наивысшее проявление иронии, язвительная открытая насмешка.
Саркастическим содержанием наполнены произведения Лермонтова, Салтыкова-Щедрина, приемами саркастического стиха блестяще владел Маяковский. Мастерству проявления сарказма учит своего мужа госпожа Москалева из «Дядюшкиного сна» Ф.М. Достоевского.
Гротеск смешивает в одном образе диаметрально противоположные понятия: фантазию и реальность, красоту и безобразность, трагедию и комедию для усиления выражения замысла автора.
В результате читатель наблюдает причудливую смесь неправдоподобных комических несоответствий, совершенный абсурд («История одного города» М. Салтыкова-Щедрина).
Риторический вопрос. Часто для усиления эмоциональной стороны речи, текста автор задается вопросом, на который не ожидает ответа (либо вопрос задан не конкретному собеседнику, либо ответить на заданный вопрос невозможно в принципе).
Риторический вопрос всегда усиливает эмоциональный «ответ» читателя (слушателя). Самый, пожалуй, известный риторический вопрос в русской литературе – слова Н.В. Гоголя: «Русь! куда же несешься ты?»
Восклицание – особая фигура стилистики речи, служащая для усиления эмоциональной стороны речи.
Восклицание может носить в одних случаях риторический характер, в других – иметь признаки гиперболизации (изречение Н.В. Гоголя о птице, не сумеющей долететь до середины Днепра – явная гипербола).
Афоризмом называется лаконичное, емкое по содержанию, краткое выражение какой-либо мысли.
Часто в литературный афоризм превращаются несколько поэтических строк, («Как мало пройдено дорог, как много сделано ошибок») либо прозаическое изречение («Любите книгу – источник знаний».
Инверсия, повторение, анафора
Литераторы постоянно ищут возможность усилить впечатление читателя, слушателя от своего творения. В ход идут различные стилистические, грамматические приемы, направленные на придание слову, тексту дополнительной выразительности. Одним из таких особых приемов является
Инверсия – перестановка слов в предложении, сознательное нарушение общепринятых правил грамматики.
Таким способом создается причудливый, необычный слог произведения, придающий тексту характерное своеобразие.
Среди остальных изобразительных средств повторение является наиболее знакомым – слова чаще всего повторяются в песнях, в стихотворных строчках.
Таким приемом автор старается усилить эмоциональное окрашивание текста, привлечь к повторению повышенное внимание.
Анафора создает повторение в тексте, строки одинаково начинаются.
Например, у Пушкина:
Блажен, кто смолоду был молод,
Блажен, кто вовремя созрел…
Повторяющееся первое слово в строке – анафора.
Кроме того, в поэтическом произведении могут повторяться группы слов, или несколько строф.
Изобразительно – выразительные средства в художественном произведении
Изобразительными средствами языка произведения художественной литературы определяют сравнение, эпитет, метафору, метонимию, гиперболу и другие средства выразительности.
Сравнение проясняет какое — либо явление/понятие путем сопоставления (сравнения) с другим предметом/явлением.
Эпитетами автор подчеркивает свойства, признаки какого-либо предмета, придает им дополнительную эмоциональную окраску. Наиболее часто для эпитета используются метафоры.
Метафора. Слово не всегда употребляется в своем прямом значении, оно может иметь также переносный смысл, как бы «взять взаймы» у другого слова или понятия его значение.
Иначе говоря, свойства одного объекта переходят на другой объект. Например, в выражении «золотая осень» желтая осенняя листва названа золотой не потому, что она растет на настоящем золотом дереве, а по сходству с золотом – металлом желтого цвета. При этом в тексте поясняющие слова (как будто, наподобие, напоминающие) отсутствуют, но всегда подразумеваются.
Метафора всегда повышает выразительность речи, усиливает эмоциональную сторону произведения. В основном выражается существительным или глаголом, реже – остальными частями речи.
Разновидности метафоры:
олицетворение
метонимия
синекдоха
Стремясь придать произведению яркую образность, автор пользуется переносом по смежности понятий – метонимией.
Существует несколько приемов подобного переноса:
Внешнее выражение и внутреннее состояние уподобляются
Действие уподобляется своему орудию
Предмет уподобляется своему хозяину
Уподобление вместившего с вмещающемся в него
Гипербола – одно из самых часто употребляемых художественных средств значительного преувеличения свойств предметов, размеров явлений, силы чувств.
Гипербола может идеализировать предмет или явление, на которое направлено, но также иметь уничтожающую направленность.
Аллегория используется для усиления выразительности поэтической речи, изображает абстрактное понятие при помощи конкретного образа.
Аллегория включает смысловой элемент (явление или понятие, изображаемое автором, которое при этом не называется), а также образно – предметный (конкретный предмет, представляющий данное явление, понятие).
Оксюморон, или оксиморон – стилистическая фигура, помогающая создать яркое образное выражение, понятие при помощи противоположных по значению слов, сочетая несочетаемое.
Например, «живой труп», «мертвые души», «старый новый год».
С этой задачей блестяще справляются ассонанс, звуковые повторы, аллитерация.
Аллитерацию чаще всего используют поэты, так как прием заключается в повторении согласных звуков, создающих оригинальную звукопись при описании картин природы, шумовое оформления текста, создающее дополнительную выразительность художественного слова.
Ассонанс является также звуковым повторением, но только уже гласных звуков.
В поэтической речи ассонансом признается неточная рифма, когда присутствует точное совпадение гласных и неточное – согласных звуков. Путем употребления ассонанса (как и аллитерации) достигается особая «зрительность» художественного образа. Ассонанс прочно присутствует в фольклоре, в классических произведениях Лермонтова («Бородино»), Блока («Фабрика»), Маяковского.
Если хотят отметить своеобразие авторского письма, индивидуальную манеру построения фразы, интонационные особенности, характерные черты языка писателя – говорят о стиле произведения.
Именно стиль придает литературному опусу оригинальность и то своеобразие, по которому читатель без труда определит, чьему перу принадлежит текст.
Дело в том, что каждый художник слова обладает ярко выраженными особенностями письма, т.н. авторской манерой.
К примеру, моментально узнается Маяковский по отточенности коротких, похожих на лозунги, строчек знаменитой «лесенки». Или супердлинные, на целый печатный лист, предложения в тексте Л.Н. Толстого.
Лаконизм и сдержанность фраз отличает писательский стиль И.С. Тургенева, обстоятельное многословие присутствует И.А. Гончаров.
Стиль может также пониматься как индивидуальная писательская манера – как строится фраза, какие изобразительные средства языка.
Мыслители, деятели культуры разных эпох и стран много рассуждали о феномене смеха в жизни человека. Из античности к нам пришло знаменитое утверждение о том, что человек отличается от животного способностью говорить и смеяться. Ф. Рабле называл смех сущностью человека. Религиозный философ В. Соловьев утверждал, что «смех предусматривает свободное состояние и волеизъявление: раб не должен смеяться». По мнению И.В. Гёте, нигде так не проявляется человеческий характер, как в его понимание и находки смешного.
Природу смешного можно уловить, обратившись к значению слова «комический». Это слово произошло от греч. Comos — так называли ватагу весельчаков, ряженых людей на сельском празднике Диониса. У всех народов известна традиция после месяцев напряженного труда и ограничений, проводить шумные праздники, карнавалы. В противовес обыденной нужде и заботам, предыдущим и грядущим будням, повседневной серьезности в этот период царили безудержное веселье, фантазия, игра. Такое времяпрепровождение воскрешало человека, восстанавливало его силы – подобно школьной перемене.
Известный ученый-филолог М. М. Бахтин, исследовавший проблему народной культуры смеха, указывал на народно-праздничные особенности характера смеха в гоголевских «Вечерах на хуторе близ Диканьки». В ранних произведениях писателя существенную роль играет «веселая чертовщина» с ее атмосферой вольности и озорства, переодеваниями и всякого рода мистификацией. Народная своеобразная основа гоголевского смеха сохранилась в творчестве писателя и в последующие годы. Элементы общественно-народной смеховой культуры мы находим в особом художественном методе Гоголя, получившем название «фантастический реализм». Хотя в «Ревизоре» нет ни одного положительного персонажа, Н. Гоголь подчеркивал, что в комедии «было одно честное, благородное лицо» — смех.
В ходе поэтапного развития культуры из народно-карнавального синтетического смеха выделились отдельные виды комического: юмор, ирония, сатира, сарказм.
У каждого вида комического свои правила игры. В иронии смешное старательно скрывают под маской серьезности, в юморе — серьезное маскируют под видом смешного. В юморе за смешным нередко прячется грустное, а вот смех саркастический имеет один эмоциональный оттенок — едко-обличительный. Сатире также присуща бескомпромиссность суждений о предмете осмеяния.
Несмотря на различия, все виды комического служат одной цели — пробуждению у человека стремления к идеалу, добру, истине, красоте. Указывая на недостойности несоответствующие определенным нормам общественной жизни, комическое способно духовно пристыжать и одновременно очищать смеющегося. В ходе поэтапного развития культуры из народно-карнавального синтетического смеха выделились отдельные виды комического: юмор, ирония, сатира, сарказм.
Юмор, ирония и сатира как моральная критика в JSTOR
Abstract
Исследуется случайная роль юмора как средства моральной критики. Я начинаю с различия между этой конкретной ролью и другими способами, с помощью которых юмор и развлечение могут рассматриваться через призму морали, рассматриваю исторические подходы к юмору, подтверждающие его роль, на которой сосредоточено мое исследование, и заканчиваю рассмотрением современные примеры иронии, сарказма и сатиры как проводников рассматриваемой критики (например, обложка New Yorker от 21 июля 2008 г.).
Информация о журнале
«Журнал эстетического воспитания» — уважаемый междисциплинарный журнал, который фокусируется на разъяснении вопросов эстетического воспитания, понимаемых в самом широком смысле. Таким образом, журнал приветствует статьи по философской эстетике и образованию, по проблемным областям образования, имеющим решающее значение для искусства и гуманитарных наук на всех институциональных уровнях; к пониманию эстетического значения новых средств коммуникации и эстетики окружающей среды; и к пониманию эстетического характера гуманистических дисциплин.Журнал — ценный ресурс не только для педагогов, но и для философов, искусствоведов и искусствоведов.
Информация об издателе
Основанная в 1918 году, University of Illinois Press (www. press.uillinois.edu) считается одной из самых крупных и выдающихся университетских издательств страны. Press публикует более 120 новых книг и 30 научных журналов каждый год по множеству предметов, включая историю Америки, историю труда, историю спорта, фольклор, еду, фильмы, американскую музыку, американскую религию, афроамериканские исследования, женские исследования и Авраама. Линкольн.The Press является одним из основателей Ассоциации прессов американских университетов, а также History Cooperative, онлайновой коллекции из более чем 20 журналов по истории.
Права и использование
Этот предмет является частью коллекции JSTOR. Условия использования см. В наших Положениях и условиях Авторское право 2011 г. Попечительским советом Иллинойского университета. Запросить разрешения
Сатира и ирония
Критические очерки
Сатира и ирония
Во многом благодаря Кандид, Вольтер считается с Джонатаном Свифтом одним из величайших сатириков в литературе.Сатиру можно определить как особый литературный способ улучшения человечества и его институтов. Сатирик критически настроен и обычно подает материал с остроумием и юмором. Осознавая серьезные ограничения в учреждениях, воздвигнутых человечеством, он может с помощью смеха стремиться произвести реконструкцию, а не разрушить их. Таким сатириком следует назвать Вольтера, и он стремился к наиболее основательному изменению человеческого поведения и институтов.
В основном сатира бывает двух видов: та, которая следует традициям Горация, мягкая, учтивая, добродушная и направленная на исправление с помощью терпимого, сочувственного смеха; и Ювенала — язвительного, оскорбительного, насмешливого и исполненного морального негодования по поводу развращенности и зла человека и его институтов. Другими словами, можно сказать, что сатира Гората развлекается безумием, а сатира Ювеналии атакует преступления или, по крайней мере, преступления, которые считаются антисоциальными.Очевидно, что последний тип, если он вообще вызывает смех, вызывает презрительный смех. Оба типа сатиры встречаются в Candide. И важно то, что даже когда Вольтер был наиболее возбужден, он использовал легкое прикосновение и достигал тона, зачастую веселого, что обманчиво для буквально мыслящего читателя, который принимает сказку как преувеличенное повествование о приключениях главного героя и не более того. . Первичный прием Вольтера как сатирика — это ирония, применяющая ее не только к высказыванию, но и к событию, ситуации и структуре.
Ирония — это риторический прием, с помощью которого реальное намерение писателя или говорящего выражается способом, имеющим противоположное значение. Нередко, как и в творчестве Вольтера, он отличается мрачным юмором. Обычно писатель устанавливает слова похвалы, чтобы выразить вину, и слова порицания, чтобы выразить похвалу, причем первая практика более распространена. Как литературный прием ирония эффективна, потому что требует сдержанности. Сатирик, который полагается на это, никогда не опускается до ругательств или сарказма; он ожидает, что его аудитория поймет суть.Можно понять, почему Тьеро восхвалял Вольтера как «превосходнейшего автора шуток и шуток» и что и барон Гримм, и мадам. Де Сталь подчеркнул комические аспекты Candide , не игнорируя при этом серьезность, лежащую в основе сказки.
Цели сатиры Вольтера многочисленны и разнообразны. Безусловно, первым по важности является философский оптимизм; другие включают религию, королей и государство, войну, алчность, социальную гордость и глупости того или иного рода. В моральном порядке нечестность, притворство, проституция и все серьезные и мелкие проявления бесчеловечности человека по отношению к человеку подвергаются нападкам, как и в естественном порядке болезни, катаклизмы и пороки развития. Для своей цели Вольтер особенно полагался на преувеличение, но он также использовал контрастирующий прием преуменьшения, часто в форме литотес, который является преуменьшением, при котором что-то утверждается путем констатации отрицания своей противоположности — распространенный прием в ироническом выражении. С ним связан эвфемизм, фигура речи, в которой косвенное утверждение заменяется прямым. Многие авторы использовали эвфемистические термины, чтобы избежать резкости или оскорбления, но они демонстрируют тенденцию к неискренности и сентиментальности.Вольтер использовал их иронично с прекрасным комическим эффектом, чтобы выразить свою сатиру на несправедливость, преступление и глупость. Также следует отметить карикатуру и пародию — способы, с помощью которых автор преувеличивал те или иные детали с той же целью.
Основная цель Вольтера при написании Candide состояла в том, чтобы опровергнуть теорию оптимизма, и для этой цели преувеличение послужило ему лучше всего. Он противопоставлял грубую абсурдность абсурду — доктрину, неоднократно озвученную Панглоссом и повторенную его учениками, против выводов, которые следует сделать из записанных фантастических опытов.Кандид изгнан из того, что для него и других в замке барона было «лучшим из всех возможных миров». Резня булгарско-абарского конфликта, буря и землетрясение, очевидная смерть Кунегонды и фактическая смерть ее родителей, события во время инквизиции — эти и все другие важные события описаны в преувеличенных выражениях.
Превосходная степень доминирует с самого начала. Жизнь в замке Thunder-ten-tronckh утопична, это жизнь совершенного счастья.Это «красивейший замок». Кандид представлен как «самый кроткий из персонажей», который сочетал в себе довольно здравое суждение с большой простотой ума. Барон — великий и могущественный лорд Вестфалии; баронесса — лучшая из всех возможных баронесс; Кунегонд — безупречная красота. Панглосс представлен как оракул, мудрейший философ в мире. Абсурд уже противопоставлен абсурду. Мы узнаем, что этот самый красивый и приятный из всех возможных замков, как Вольтер называет его в последнем предложении главы, достаточно груб, что касается его одной двери, окна и одного гобелена.Баронесса страдает ожирением; барон явно примитивный персонаж. Но все эти преувеличения, все превосходные степени готовят читателя к ужасным событиям, которые должны последовать за этим. Точно так же в описании этой неизбывной земли, Эльдорадо, и в описании лесной резиденции дона Иссахара с ее просторными садами и великолепными убранствами Вольтер снова использовал преувеличение как прелюдию к неудачам.
Автор использовал различные формы противодействия Оптимизму. Формула «лучший из возможных миров» появляется снова и снова, но ее опровергают сатирической и иронической язвой.Одна из этих форм — это своего рода преуменьшение. Кандид — хозяин этого — случайно. Часто, пережив ужасную опасность и страдания, его немедленной реакцией является то, что доктор Панглосс может — даже возможно — начать сомневаться в своей собственной философии. Выслушав рассказ старухи во всех ее ужасных подробностях, он замечает: «Очень жаль, что мудрый Панглос был повешен, вопреки обычаю, в аутодафе: он рассказал бы нам замечательные вещи о физическом и физическом. моральное зло, которое покрывает землю, и я почувствовал бы себя достаточно сильным, чтобы отважиться на несколько уважительных возражений.Вспомните также его немедленную реакцию, когда он узнал, что орейлоны, считая его иезуитом, намеревались поджарить или сварить его, а затем съесть: «Все хорошо, я не буду спорить; но я должен признать, что это жестокая участь — потерять леди Кунегонд, а затем быть зажаренной на вертеле Орейллонами ». В гостинице в испанской деревне старуха выразила свое убеждение, что отец-францисканец украл деньги и драгоценности Кунегонда. Кандид заметил, что ему следовало оставить им достаточно, чтобы завершить их путешествие.Есть ироническое преуменьшение в описании проигрышей Кандид в карты в Париже. Юноша был озадачен, потому что у него никогда не было тузов, но, как писал Вольтер, Мартин не был удивлен. Часто именно такими лаконичными высказываниями автор добивается остроумной преуменьшенности.
Вольтер имел естественную склонность к эвфемизму, и примеры этого риторического приема многочисленны в Candide. Доктор Панглосс был неизбежно эвфемистом, когда он озвучивал клише оптимизма, чтобы доказать, что даже большое зло ведет к добру.В вопросах, касающихся церкви и государства, эвфемистическое клише также служило целям Вольтера. Отчет инквизиции, например, предоставил ему прекрасные возможности для сатирических, эвфемистических комментариев. Следует вспомнить почти церемониальную вежливость темнокожего инквизитора, когда он осведомился о взглядах Панглосса в конце главы V. Бедственное положение Панглосса и Кандид было описано не менее церемонно (глава VI).
«Их разделили и поместили в очень прохладную комнату, где солнце никого не беспокоило.Через неделю они оба были одеты в санбенито и бумажные митры. В таком наряде они прошли процессией и услышали волнующую проповедь, сопровождаемую красивой полифонической музыкой. Кандид был выпорот до пения, Бискайянин и двое мужчин, отказавшихся от свинины, были сожжены, а Панглосса повесили, хотя это было не принято ».
Несоответствие этой сцены подчеркивается формальными эвфемистическими терминами, в которых она описана, и, таким образом, со ссылкой на опыт Кунегонда в качестве зрителя в инквизиции (Глава VIII).Великий инквизитор, чья незаконная страсть к ней пробудилась, когда он увидел ее на мессе, оказал ей «честь пригласить ее на службу. У нее было очень хорошее место» и наслаждался закусками, которые подавали дамам между мессой и казнями. . Как если бы она была на модном светском мероприятии!
Восторженная похвала Какамбо правительству иезуитов в Парагвае — еще один пример. «Их правительство — замечательная вещь. Королевство уже более семисот пятидесяти миль в поперечнике и разделено на тридцать провинций.У отцов есть все, у людей — ничего; это шедевр разума и справедливости. Я не знаю никого более божественного, чем отцы ».
Булгарско-абарский конфликт дал Вольтеру не меньшую возможность для сатиры, в которой он максимально использовал эвфемизм (глава II). Завербовав Кандида на службу к «самому очаровательному из королей», один из сержантов-вербовщиков сказал: «Теперь ты опора, покровитель, защитник и герой булгар: твое состояние сколочено, и твоя слава — уверен.«Сразу после этой громкой речи Кандид был закован в кандалы и отправлен в полк. Во всем этом эпизоде изобилует эвфемизмом, противоположным реальности. Мы читаем о веселой форме, волнующей музыке — и узнаем мрачные факты войны ( Глава III):
«Ничто не могло быть более великолепным, блестящим, умным или упорядоченным, чем две армии. Трубы, гобои, барабаны и пушки производили гармонию, равной которой никогда не было в аду. , затем ружейный огонь удалил из лучшего из миров около девяти или десяти тысяч негодяев, наводнивших его поверхность.«
После ужасной бойни в каждом лагере пели Te Deums: все свойства тщательно соблюдались — так Вольтер видел «славную войну».
Заслуживает внимания еще одно довольно забавное и эффективное использование эвфемизма. В первой главе прекрасный и невинный Кунегонд наблюдал за Панглосом и Пакеттом в весьма опасной ситуации. Вольтер старался не называть вещи своими именами:
«Однажды, когда Кунегонд гуляла возле замка в небольшом лесу, известном как« парк », она увидела в кустах доктора Панглосса, который проводил урок экспериментальной физики горничной своей матери, очень красивой и послушной маленькой брюнетке.Поскольку леди Кунегонд глубоко интересовалась наукой, она затаив дыхание наблюдала за повторяющимися экспериментами, которые проводились у нее на глазах. Она ясно видела достаточную причину доктора и действие причины и следствия ».
Преувеличение, преуменьшение и эвфемизм, очевидно, поддаются карикатуре и пародии, на что мы сейчас обращаем особое внимание. Яркая карикатура очевидна в описаниях, пусть кратких, барона и баронессы в главе I.Ученый доктор Панглосс, ранний и поздний, представляет собой примечательную карикатуру — как и барон-иезуит, с его заявлениями о бессмертной преданности, а затем и с его полной неприязнью. Губернатор Буэнос-Айреса со своими многочисленными именами и невыносимой гордостью стал еще одним примером. Весь обескураживающий эффект в главе I с ее контрастом наивности и догматизма — чистая пародия, особенно ложная трагедия изгнания Кандида из замка.
Ранее упоминались романтические произведения семнадцатого и восемнадцатого веков, особенно пасторальные романы и героически-галантные приключенческие рассказы, большинство из которых почти бесконечны.Вольтер, который не мог переварить их не больше, чем его прославленные предшественники, Мольер и Буало, возражал как против стиля, так и против содержания, как он ясно дал понять в своем Siècle de Louis XIV. Что касается стиля, то основными отклонениями от него были préciosité. История l’esprit precjeux была поиском элегантности и отличия в манерах, стиле и языке. Его приверженцы искали новые выражения, особенно метафорические; они избегали низких или варварских терминов и — к их большой чести — стремились к ясности и точности.В лучшем случае préciosité означало чувствительность вкуса и чувств. Но он имел тенденцию к сужению, и стиль типичного писателя-романтика легко приводил к излишествам. Страницы их произведений были заполнены красноречивыми заявлениями о бессмертной любви, потоками слез, падающими в обморок героинями, сценами внезапного узнавания, насильственной смертью, путешествиями из одной страны в другую. В Candide Вольтер, несомненно, наслаждался пародированием жанра : его герой путешествовал вдоль и поперек; Панглосса повесили, но он выжил; Сообщалось, что Кунегонд была выпотрошена, но она появилась снова; были гибель противников и бегство во временное укрытие.Экстравагантный дискурс иезуитского барона, возможно, лучше всего иллюстрирует карикатуру и пародию в повествовании. Когда он впервые встретился с Кандидом в Парагвае и узнал, кем является юноша, он был очень вспыльчивым. «Барон не уставал обнимать Кандид», — читаем мы. И тут же следует разворот. Когда барон узнал, что юноша женится на его сестре, его настроение изменилось, но его разговоры и действия были не менее резкими: «Ты, наглый негодяй! Как нагло с твоей стороны даже думать о женитьбе на моей сестре, у которой семьдесят два поколения. благородства позади нее! » Позже, когда Кандид и барон, которого Кандид на самом деле не убивал, встретились снова, барон сказал: «Ты можешь убить меня снова, но ты никогда не женишься на моей сестре, пока я еще жив».»Чтобы полностью оценить экстравагантность его слов, нужно вспомнить все, что случилось как с бароном, так и с его сестрой — одна теперь женщина, красота которой полностью поблекла в результате ее страданий, а другая только что спасла Кандид из камбузы.
Примеры и определение сатиры
Определение сатиры
Сатира — это литературный прием для искусного высмеивания глупости или порока как средство их разоблачения или исправления. Предметом сатиры обычно является человеческая слабость, поскольку она проявляется в поведении или идеях людей, а также в общественных институтах или других творениях.Сатира использует тоны веселья, презрения, презрения или негодования по отношению к ошибочному предмету с надеждой на осознание и последующее изменение.
Например, одно из самых известных сатирических литературных произведений — О дивный новый мир Олдоса Хаксли. В своем романе Хаксли высмеивает большинство социальных условностей и институтов, считающихся священными и дорогими «просвещенному» западному обществу. Это включает религию, моногамию, социальное равенство и благословение деторождения.В романе эти условности и институты перевернуты, так что персонажи охватывают культуру наркотиков, разделение на социальные классы, случайный секс и правительственный контроль. Хаксли высмеивает современное общество, чтобы показать читателю его произвольные и часто лицемерные моральные структуры.
Общие примеры сатиры
Многие распространенные формы средств массовой информации, искусства и развлечений отражают сатиру, включая фильмы, журналы, газеты, романы, поэзию, короткометражные художественные произведения, драму и даже изобразительное искусство.Сатира может быть открытой или тонкой, но она широко распространена на протяжении всей истории и в популярной культуре. Вот несколько распространенных и знакомых примеров сатиры:
политические карикатуры — высмеивают политические события и / или политиков
The Onion — американские цифровые СМИ и газетная компания, высмеивающая повседневные новости на международном, национальном и местном уровнях
Family Guy — анимационный сериал, высмеивающий американское общество среднего класса и его условности.
The Colbert Report — комедийный телесериал, высмеивающий новости и программы ночных ток-шоу.
Алиса в стране чудес j — роман Льюиса Кэрролла высмеивает коррумпированную политическую и судебную систему викторианской Англии.
Важность серьезности — драматическая сатира Оскара Уайльда о культурных нормах любви и брака в викторианскую эпоху
Шрек — фильм, высмеивающий сказки
Фонтан — знаменитый писсуар Марселя Дюшана, высмеивающий американский авангард e art
Ответ нимфы пастырю — стихотворение сэра Уолтера Рэли, высмеивающее пастырское предание Кристофера Марлоу «Страстный пастырь своей любви»
2BR02B — краткий рассказ Курта Воннегута, высмеивающий смысл жизни, смерти, и индивидуальность
Mad Magazine — сатиризированная поп-культура и политика
Deadpool — фильм, высмеивающий жанр супергероев
Скромное предложение (для предотвращения того, чтобы дети бедных людей были изгоями из-за своих родителей или страны, И для того, чтобы сделать их полезными для обществаk) — эссе Джонатана Свифта, высмеивающего правовую и экономическую эксплуатацию Ирландии Англией XVIII века
Крик — фильм, высмеивающий жанр ужасов
Mr.Робинсон — персонаж, которого играет Эдди Мерфи, высмеивающий мистера Роджерса и его детскую телевизионную программу
Примеры сатирических телевизионных программ
Многие телевизионные программы основаны на сатире. Они обращаются к аудитории своим сочетанием внимательности, юмора и критики политики, популярной культуры, социальных условностей, человеческой природы, средств массовой информации и даже самого телевидения. Вот несколько примеров сатирических телевизионных программ:
The Daily Show
South Park
The Office
Flying Circus Монти Пайтона
Saturday Night Live
The Simpsons
The Soup
American Dad
Last Week Сегодня вечером с Джоном Оливером
Женат, имеет детей
Известные примеры цитат о сатире
Один из способов лучше понять мастерство, цель и эффект сатиры — это слова самих сатириков.Вот несколько известных цитат о сатире:
Сатира — это трагедия плюс время. Вы дадите этому достаточно времени, публика, рецензенты позволят вам высмеять это. Если подумать, это довольно нелепо. (Ленни Брюс)
Завтра — это сатира на сегодняшний день, которая показывает его слабость. (Эдвард Янг)
Сатира — это урок, пародия — это игра. (Владимир Набоков)
Сатиру нельзя спорить. Вы либо понимаете, либо нет. (Майкл Мур)
Я целюсь только в сильных мира сего.Когда сатира направлена на бессильных, она не только жестока, но и вульгарна. (Молли Айвинс)
Я никогда не хотел заниматься политической сатирой, потому что мне она кажется слишком поверхностной. (Трейси Уллман)
Люди говорят, что сатира мертва. Он не мертв; он жив и живет в Белом доме. (Робин Уильямс)
Незаслуженная похвала — это замаскированная сатира. (Александр Поуп)
Сатира — это форма социального контроля, это то, что вы делаете.Это не личное. Это работа. (Гарри Трюдо)
Разница между сатирой и пародией
Некоторым бывает трудно отличить сатиру от пародии. Оба устройства используют юмор для передачи смысла и достижения своей цели. Однако между ними есть различия, особенно в их намерениях. Сатира направлена на высмеивание человеческих и / или социальных недостатков, несоответствий и несоответствий как средство провокации аудитории и оспаривания точек зрения. Пародия намеревается имитировать что-то знакомое аудитории в качестве средства развлечения или юмора.
Пародия в первую очередь полагается на признание публикой того, что имитируется, чтобы понять насмешку над объектом. Однако в центре внимания пародии обычно находится преувеличение или наблюдение на поверхностном уровне, например манеры или манера речи известного лидера. Мотив пародии — вызвать смех, а не более глубокое понимание.
Фокус сатиры — больший размах. Сатира полагается на признание аудиторией системной проблемы, лежащей в основе насмешек и юмора.Следовательно, хотя сатира действительно предназначена быть юмористической, ее мотив — это более общее понимание человечества и общества, а не смех.
Написание сатиры
В целом, как литературный прием, сатира функционирует как средство передачи социальных комментариев и / или критики со стороны писателя с помощью иронии, юмора, преувеличения и других методов. Это эффективно для читателей, поскольку сатира может создать через литературу критическую линзу, с помощью которой можно взглянуть на человеческое поведение, политические структуры, социальные институты и даже культурные традиции.
Важно, чтобы писатели помнили, что их аудитория должна понимать исходный материал, который высмеивается. В противном случае сатирический смысл теряется и оказывается неэффективным. Поэтому лучше всего осознавать способность читателя различать, какие элементы человеческой природы, истории, опыта или культуры высмеиваются в литературном произведении.
Вот несколько способов, которыми писатели извлекают выгоду из включения сатиры в свою работу:
Создавайте осведомленность и призывайте к действию в читателе
Поскольку основная цель сатиры в литературе — передать социальные комментарии и / или критику, это позволяет писателю для повышения осведомленности о проблемах и неравенствах в обществе. Сатирическая литература привлекает внимание к этим вопросам и может рассказать читателям о том, чего они раньше не рассматривали или не понимали. Это осознание может затем вызвать у читателя призыв к действию, чтобы он осудил, попытался исправить или даже более критически подумать о социальных недостатках.
Создайте сочувствие и рефлексию для читателя
Многие писатели считают сатиру литературным приемом, который позволяет им подносить своему читателю метафорическое зеркало. Это позволяет читателю сопереживать обездоленным в сатирических произведениях, а также дает возможность поразмышлять о собственном поведении и / или точке зрения читателя.Другими словами, если сатира в литературе относится к поведению или мировоззрению читателя, то он может задуматься о своем соучастии.
Примеры сатиры в литературе
Сатира — очень эффективное литературное средство, способное изобразить и отразить социальные комментарии и критику. Вот несколько примеров сатиры и того, как она увеличивает значимость известных литературных произведений:
Пример 1:
Lysistrata (Aristophanes)
LYSISTRATA: Пусть нежная Любовь и милая Кипрская королева осыпают соблазнительными чарами на нас. пазухи и все наше лицо.Если только мы сможем вызвать у людей такое любовное чувство, чтобы они стояли твердо, как палки, мы действительно заслужили бы имя миротворцев у греков.
В этой греческой комедии поэт Аристофан создает главную героиню, Лисистратту, которая убеждает своих собратьев воздерживаться от всех сексуальных взаимодействий со своими партнерами-мужчинами, чтобы повлиять на них и заставить их прекратить Пелопонезийскую войну. В пьесе Аристофан высмеивает войну, но вместе с тем высмеивает сложности взаимоотношений между мужчиной и женщиной и подразумеваемый характер различий между мужчинами и женщинами.История Лисистраты продолжала адаптироваться и интерпретироваться с течением времени, что указывает на то, что комедийные темы оригинала остаются кормом для сатиры.
Пример 2:
Сон в летнюю ночь (Уильям Шекспир)
Ay me! насколько я мог когда-либо прочесть, Могу когда-либо услышать по сказке или истории, Истинная любовь никогда не шла гладко
Это утверждение Лисандра в пьесе отражает умное использование Шекспиром сатиры как литературного приема.Фактически, в основе этой комедийной пьесы лежит сатира о том, как люди по глупости воспринимают и идеализируют понятие романтической любви. Персонаж Лисандра отражает эту иронию, показывая, что он никогда не слышал и не читал любовную историю, которая не была бы неприятной. Поэтому идея о том, что персонажи пьесы поглощены романтическим представлением о любви, иррациональна, учитывая, что нет основополагающего примера успешной или «гладкой» страстной любви, на которой можно было бы основывать их идеализацию. Шекспир высмеивает этот тип любви, высмеивая глупое поведение людей во имя романтики и страсти.
Пример 3:
Несчастное совпадение (Дороти Паркер)
К тому времени, когда вы клянетесь, что вы его,
Дрожа и вздохи,
И он клянется, что его страсть
Бесконечная, бессмертная — —
Леди , запишите это:
Один из вас лжет.
Дороти Паркер — одна из самых известных и успешных сатириков. Ее стихи часто обращаются к теме любви с художественной композицией, однако она постоянно использует свой талант юмора и сатиры, чтобы высмеивать жанр романтической поэзии и сам предмет любви.Это видно из ее стихотворения «Несчастное совпадение», в котором она изображает двух влюбленных, которые заявили о своей вечной любви и страсти друг к другу. Вместо того, чтобы праздновать этот роман, Паркер высмеивает его, предупреждая «леди» в стихотворении, что либо она, либо ее возлюбленный лгут.
Сатира Паркер на романтическую любовь привлекает внимание читателя к частым ложным надеждам и обещаниям романтической любви, любовников и даже романтических стихов. Это позволяет читателю оценить художественный характер любовного стихотворения, одновременно придя к пониманию того, что концепция романтической любви не является устойчивой и является ложной реальностью.
Сатира против пародии
Сатира и пародия — это одно и то же? Даже писателям легко предположить, что они похожи друг на друга. В конце концов, оба используют юмор, чтобы подшутить над объектом насмешек или насмешек, поэтому у них, безусловно, есть общий импульс. Тем не менее, они не идентичны, поэтому знание разницы между сатирой и пародией является ключевым моментом для любого начинающего писателя. Тем, кто хочет писать с юмором, полезно узнать, как они сосуществуют и иногда переплетаются, чтобы помочь вам прибегнуть к выбору наиболее подходящей техники в зависимости от вашего настроения.Давайте начнем с нескольких простых определений, чтобы их различать.
Сатира
Сатира использует юмор (особенно иронию и преувеличение), чтобы выявить недостатки в человеческом поведении. По большому счету, любой, кто пишет сатирический рассказ, намеревается высмеять идиотизм или пороки людей. Когда представления о человеческой слабости, непристойности или неадекватности сопоставляются с другими факторами, такими как социальные проблемы или политические комментарии, сатира может стать мощным инструментом, чтобы спровоцировать и бросить вызов установкам.Часто именно благодаря использованию юмора сатирические произведения могут быть очень похожи на предвестники перемен, помогая сформировать общественное мнение вокруг единого понимания.
пародия
Пародия — это любое произведение, имитирующее знакомый стиль (художника, жанра или произведения), чтобы вызвать юмор. Как и сатира, пародия полагается на преувеличение, чтобы высмеять свою цель, но ее основная цель — развлечься, подражая тому, что другие могут узнать. Однако помимо этого есть немного более глубоких мотивов. Большинство пародистов сосредоточено на высмеивании поверхностных наблюдений ради легкого (если не заслуживающего) смеха, как, например, книга Роберта Сирса «Прекрасная поэзия Дональда Трампа». Это может показаться сатирой, но это не значит, что это так.
Разница
Я бы сказал, что пародию лучше всего рассматривать как путь к сатире, или своего рода легкое ее факсимиле. В то время как сатира сфокусирована на более широкой картине — намереваясь высмеять более глубокие проблемы, выходящие за рамки избранного литературного стиля, — пародия во многих случаях может быть поверхностным примером мимикрии, которая не имеет длительного воздействия (например, «Скучающие кольца» Генри Бирда и Дугласа Кенни). Тем, кто любит посмеяться, оба стиля могут что-то предложить, и сходство между сатирой и пародией, безусловно, остается незаметным, но, по моему личному мнению, пародия — это кирпич, а сатира — стена.
Все сводится к тому, чтобы использовать свои сильные стороны как писателя — если вас больше интересуют проблемы, связанные с человеческой природой, например, как Джордж Оруэлл был в «Скотной ферме» и «Девятнадцать восемьдесят четыре», то написать сатиру было бы более важным. — всеобъемлющий, жизненно важный и проницательный для ваших читателей. Если все, что вы хотите сделать, это высмеять или убрать микки из чего-то конкретного, что вас раздражает, например, из фильма или телешоу, то пародия, возможно, является лучшим средством достижения этого, но я бы не ожидал, что это будет иметь особенно долгий срок хранения.Нельзя сказать, что сатира не может иметь пародийных элементов или эта пародия не может иметь сатирических качеств, но амбиции, которые в конечном итоге разделяют их. Писатели должны сами определять, какой метод выбрать, когда приходит вдохновение.
Люк Эдли
Юмористический писатель-фантаст, поэт и прозаик. Увлекается сатирой. Интересуюсь юмористическими романами, черной комедией и сказками о сатирических безрассудках.
Понимание различных типов сатиры
Сатира существует уже тысячи лет, поэтому неизбежно, что на протяжении всей своей эволюции она как литературный жанр приобрела множество сложностей. Можно утверждать, что три наиболее распространенных типа сатиры (хоратовская, ювеналийская и мениппийская) теперь смешались и перекрестно опыляются до такой степени, что нередко современное сатирическое произведение представляет собой своего рода гибридную дворнягу.
Однако, если вы писатель, планирующий придать своей истории сатирический оттенок, важно понимать, каковы ключевые составляющие различных типов сатиры. В наши дни писатели могут позволить себе отбирать элементы одного и переходить к другому в рамках одного повествования, но все же важно знать различия.Изучение ключевых особенностей разных направлений сатиры, несомненно, поможет вам выбрать направление.
Вот краткий учебник по трем наиболее распространенным типам сатиры, который поможет вам начать работу.
1. Сатира Горациана
Скорее всего, если ваша цель — только рассмешить людей, вам нужна хоратовская сатира. Названный в честь римского сатирика Горация, который начал писать сатирические стихи в 35 г. до н.э., его цель заключалась в основном в том, чтобы развлечься кривым юмором, остроумием и беззаботной насмешкой, избегая негатива, отказываясь возлагать вину на других за любые предполагаемые опасения.Таким образом, цель сатиры Гората — быть умным и знающим, вызывая при этом юмор, раскрывая особенности человеческого поведения.
«Фарс» или «комедия ошибок» могут, например, иметь хоратовское чутье, но нередко горатовские сатиры прибегают к высмеиванию преобладающих социальных установок (таких как проделывание дыр в философских позициях и социальных нормах). То, как брак и отношения воспринимаются в романе Джейн Остин «Гордость и предубеждение», было бы примером сатиры Гората, равно как и фарсовая социальная активность и легкое остроумие, содержащиеся в «Важности серьезности» Оскара Уайльда.
Тем не менее, хотя хоратовская сатира и юмористична, она является самой мягкой и нежной формой сатиры из всех существующих — она не стремится изменить мир. Он просто сосредоточен на освещении человеческого безумия во всех его бесчисленных формах, возможно, посредством анекдотов и характеристик, а не сюжета, и поэтому его главная цель — в первую очередь развлекать.
2. Ювенальная сатира
Если гнев — это ваша энергия — например, если вы хотите ниспровергнуть статус-кво и атаковать продажность политического класса или религиозных лидеров, — тогда ювенальская сатира — ваш лучший выбор.Миссия ювенальной сатиры, освобожденной от оков откровенного веселья, часто состоит в том, чтобы нападать на людей, правительства и организации с целью разоблачения лицемерия и моральных проступков. По этой причине писателям следует ожидать, что они будут использовать более сильные дозы иронии и сарказма в этой выдумке.
Возникший у римского сатирика Ювенала в конце I века до нашей эры, горькая резкость, которую он привносил в свои работы — с почти презрительным, резким, обвиняющим и кричащим рвением, — повлияла на сатирическую разновидность, подарившую нам самую резкую и провокационную. и памятные литературные произведения.«Путешествие Гулливера» Джонатана Свифта является ярким примером этого, главным образом в том, что касается абсурда и лицемерия политики и религии. Проще говоря, если ваша история подпитывается моральным негодованием или личными оскорблениями, вы обнаружите, что попадаете на территорию ювеналии.
Неудивительно, что Джордж Оруэлл также очень часто увлекался ювеналийской сатирой в своих романах (особенно «Скотный двор» и «Девятнадцать восемьдесят четыре»). Поэтому, если у вас есть сильная сторона, которую нужно указать, и явная цель для вашего презрения, этот тип сатиры будет наиболее эффективным, хотя часто пессимистичным и пронизанным горечью.В результате ювенальская сатира часто не такая юмористическая, как другие виды сатиры, но, возможно, она наиболее смелая и революционная.
3. Менипповая сатира
Вместо того, чтобы сосредотачиваться на социальных нормах, менипповая сатира имеет тенденцию высмеивать индивидуальный недостаток характера и / или определенную черту личности, такую как умственное отношение. Думайте об этом как о чуть более острой версии сатиры Гората, в которой он атакует конкретный человеческий недостаток, а не очевидный проступок. Например, «Алиса в стране чудес» Льюиса Кэрролла — это менипповая сатира в том смысле, что именно любопытство Алисы в конечном итоге приводит к ее тяжелому положению.
Получив свое название от греческого философа Мениппуса III века до нашей эры, этот тип сатиры гораздо менее агрессивен, чем ювенальская сатира, но заметно более критичен, чем его горатский кузен. Именно здесь вы увидите, например, высмеивание сексистских взглядов или расистских взглядов, напыщенности или высокомерия (среди бесчисленных других человеческих недостатков в более общем плане).Короче говоря, любая точка зрения или отношение, делающие человека достойным высмеивания, может стать мишенью для менипповой сатиры.
Судя по манере, в которой менипповая сатира выражает моральные суждения, может показаться, что она сделана из той же ткани, что и ювеналийская сказка, но она не должна быть жесткой и может быть такой же легкой и даже такой же веселой, как сатира Гората. Как читатель, человек должен уметь признать, что причина веселья в Менипповой сатире, в конечном счете, заключается в изображении взглядов персонажей, высмеивании пороков, связанных с конкретным умственным недостатком или врожденной слабостью личности.Это главное, что отличает его от других видов сатиры.
Что объединяет каждый тип сатиры, так это склонность использовать иронию, сарказм, юмор и насмешки, чтобы позволить писателю достичь своих целей. Возможно, именно поэтому каждый исторический тип сатиры со временем сливается и накладывается друг на друга — в конце концов, если методы одинаковы, трудно избежать достижения одного и того же пункта назначения. Надеюсь, однако, что понимание того, что делает каждый тип сатиры уникальным, даст писателям инструменты, необходимые для экспериментов и сохранения этой литературной традиции.Пусть это будет продолжаться долго.
Синонимов иронии | Тезаурус Мерриам-Вебстера
ситуация, которая является странной или забавной, потому что события происходят не так, как ожидалось
Мы получили хороший смешок, увидев, что их эвакуатор отбуксируют, но наши друзья не смогли увидеть иронию .
комедия,
комикс,
комичность,
дроллер,
забавность,
забавность,
веселье,
юмор,
юмористика,
богатство,
шумность
См. Определение словаря
Что такое сатира — 3 типа сатиры, которые должен знать каждый писатель
определение сатиры
Что означает сатира?
Что означает сатира? Это термин, который вы часто слышите, но есть разные типы сатиры с разными подходами и результатами.Первый шаг к пониманию сатирического — это понять, что такое сатира в общих чертах. Для этого давайте взглянем на определение сатиры.
ОПРЕДЕЛЕНИЕ САТИРА
Что такое сатира?
Сатира — это жанр, в котором преувеличение, ирония, юмор или насмешки используются для критики и выявления недостатков в человеческой природе и поведении. Помимо того, что это отдельный жанр, это литературный прием, который часто используется для критики политики и злободневных вопросов.
Сатира используется в различных средствах массовой информации, таких как кино, литература и даже музыка. Цель сатиры — развлечь публику и побудить ее глубже задуматься о предмете. Часто это шутка, но не обязательно.
Слово сатира происходит от латинского словосочетания «satur», означающего «полный», и фразы «lanx satura», что буквально переводится как «блюдо, полное различных фруктов». Хотя это может показаться далеким от его современного значения, термин «сатира» использовался римскими критиками.Источником этого жанра считается «Старая комедия » Аристофана « y ».
Чем создается сатира?
Для чего используется сатира?
Типы сатиры
Сатира — невероятный инструмент для критики человеческой природы посредством творчества. Есть три разных типа сатиры, которые используют сценаристы в зависимости от того, как они хотят ее использовать. Вот профессор Готтлиб из Университета штата Орегон с обзором этого жанра и его функции.
Что такое сатира?
1. Горатовская сатира
Горатовская сатира, пожалуй, самый распространенный вид сатиры. Горатовская сатира обычно использует юмор, чтобы высмеять человека или событие в комедийной манере. Цель здесь — быть беззаботным и средством поощрения улучшения того, что высмеивается. В этом типе часто встречается использование пародии.
Горатовская сатира обычно используется для беззаботного социального комментария.Сегодня такие шоу, как The Colbert Report и Saturday Night Live , используют эту сатирическую стратегию.
«Доктор Стрейнджлав» или «Как я научился перестать волноваться и полюбил бомбу» , один из лучших фильмов Стэнли Кубрика, использует хоратовский стиль, чтобы дать забавные комментарии к политике холодной войны. Одна из самых знаковых строк фильма прекрасно отражает блестящее использование Кубриком этой техники.
Доктор Стрейнджлав • Прочитать всю сцену
Конечно, не все сатирические материалы беззаботны и юмористичны.Для чего используется сатира, если она не смешит аудиторию или читателя? Давайте посмотрим на более темный шрифт.
2. Ювенальная сатира
Ювенальная сатира, в отличие от Горациана, намного мрачнее и серьезнее. Он часто используется, чтобы выразить разочарование или гнев по поводу текущего состояния. Этот тип, обычно встречающийся в жанре антиутопии, направлен на выявление недостатков политических и культурных систем и часто вызывает споры из-за отсутствия сдержанности.
Прекрасным литературным примером этого типа является роман Джорджа Оруэлла « 1984 ».Роман использует политическую сатиру, чтобы привлечь внимание к недостаткам и опасностям тоталитарного правительства. Другие классические романы, такие как Fahrenheit 451 и A Clockwork Ora nge, также используют эту технику, чтобы выявить существующие проблемы.
3. Менипповая сатира
Что такое сатира, которая критикует общие системы убеждений, а не человека или отдельного человека? Это не что иное, как менипповая сатира. Возможно, вы не знаете определение менипповой сатиры, но вы определенно видели его по телевидению.
Этот подход неоднократно использовался в телешоу South Park для высмеивания таких тем, как расизм, классизм, гомофобия и ксенофобия. Мениппейский стиль часто требует большей продолжительности работы, чтобы создать свою точку зрения.
Литература по определению сатиры
Сатира в литературе
Для чего сатира используется в литературе? Для авторов это инструмент, который позволяет авторам оставлять комментарии в социальных сетях. Хотя многие сатирические романы изначально давали представление об обществе во время написания романа, иногда его актуальность может относиться к обществу сегодня.Прекрасным примером этого является роман Ральфа Эллисона « Человек-невидимка» .
В книге Эллисон использует сатирический подход, чтобы комментировать социальные проблемы и расовую несправедливость, с которыми афроамериканцы столкнулись в двадцатом веке. Хотя роман был написан в 1952 году, он стал более важным 12 лет спустя во время движения за гражданские права.
Сегодня роман по-прежнему актуален и так же важен, как и тогда, когда он был впервые написан из-за расовой несправедливости в текущих событиях.Вы можете узнать больше о Invisible Man и его влиянии на видео ускоренного курса ниже.
Литература по определению сатиры • Человек-невидимка
Человек-невидимка — не единственный роман, критикующий социальную несправедливость и абсурдность. Другие примеры сатиры в романах, таких как « Animal Farm » Джорджа Оруэлла, «Бойня № 5» Курта Воннегута и «Бесконечная шутка » Дэвида Фостера Уоллеса, создают мастерские истории с использованием сатирического содержания. Другой распространенный в литературе тип инструмента — пародия. Так что же означает сатира по сравнению с пародией?
Разница между сатирой и пародией
Сатира и пародия
Пародия и сатира часто ошибочно принимают друг за друга, и это справедливо. Оба используют одинаковые литературные приемы, такие как преувеличение, юмор и ирония для конечного результата. Они различаются по мотивам и масштабам.
Сатира использует эти литературные приемы, чтобы привлечь внимание к недостаткам в человеческой природе и поведении.Его мотив — побудить аудиторию глубже задуматься над темой и, возможно, изменить свое отношение или мнение о ней.
Сатирический новостной сайт « The Onion » содержит отличные сатирические примеры того, как они направлены на критику человеческого поведения. Взгляните на одно из их видео ниже, которое высмеивает потребительство и чувство превосходства над Wal-Mart в этой фальшивой новости.
Примеры сатиры • The Onion
Parody, с другой стороны, стремится имитировать стиль или форму чего-то или кого-то еще (например, художника или фильма) для создания юмора.Пародия использует то, что знакомо публике, и сочетает это с преувеличением, иронией и юмором, чтобы развлечь их.
Действительно, многие пародии направлены на издевательство над политиками и знаменитостями, что может быть сатирическим. Однако не все пародии — сатиры.
Например, возьмите эту пародию на фильм 1917 под названием 2020 от Ascender.
НЕСОБСТВЕННЫЙ ИНТЕГРАЛ — это… Что такое НЕСОБСТВЕННЫЙ ИНТЕГРАЛ?
— интеграл от неограниченной функции или от функции по неограниченному множеству. Пусть функция f определена на конечном или бесконечном полуинтервале , и для любого функция f интегрируема но Риману (по Лебегу) на отрезке Тогда предел
(в случае условие понимается как ) наз. несобственным интегралом
Если предел (1) существует, то говорят, что Н. и. сходится, если не существует — расходится. Напр., Н. и.при сходится, а при расходится.
Если же , то
сходится при и расходится при .
Если и функция f интегрируема по Риману (по Лебегу) на отрезке [ а, b], то Н. и. (1) совпадает с определенным интегралом.
Аналогично при соответствующих предположениях определяют Н. и. по промежутку
Если функция/ интегрируема по Риману (по Лебегу) на каждом отрезке и существуют то Н. и.
определяется как сумма
и не зависит от выбора точки с.
Если на интервале ( а, b )имеется конечное число точек
: таких, что функция f интегрируема по Риману (по Лебегу) на каждом отрезке , не содержащем ни одной точки , и для каждого существуют Н. и.
то Н. и.
Это определение не зависит от выбора точек .
На Н. и. переносятся общие свойства интегралов: линейность, аддитивность относительно промежутков, по к-рым производится интегрирование, правило интегрирования неравенств, теоремы о среднем, интегрирование по частям и замены переменного, формула Ньютона — Лейбница. Напр., если функция f почти всюду на [ а, b )совпадает с производной функции F, к-рая абсолютно непрерывна на каждом отрезке то
Для выяснения сходимости Н. и. от знакопостоянных функций применяется признак сравнения: напр., для Н. и. вида (1) при выполнении условия
из сходимости Н. и.
следует сходимость Н. и.
функция наз. в этом случае функцией сравнения. В качестве функции сравнения для интегралов (1) в случае конечного предела интегрирования bчасто используются функции ; для интегралов вида (2) в случае конечности предела интегрирования а- функции , при наличии одного или двух бесконечных пределов интегрирования — функции . Из признака сравнения следует, напр., если для неотрицательной функции f, определенной при , существует предел
то при Н. и.
вида (1) сходится, а при Н. и. расходится.
Необходимое и достаточное условие сходимости Н. и. дает критерий Коши. Так, Н. и. вида (1) сходится тогда и только тогда, когда для любого существует такое что для всех выполняется неравенство
—
Н.
наз. абсолютно сходящимся, если сходится Н. и.
Если Н. и, абсолютно сходится, то он сходится и совпадает с интегралом Лебега. Существуют Н.
а для бесконечного:
Существуют различные признаки для установления сходимости Н. и. Так, если функции f и gопределены для , функция f имеет на полуоси ограниченную первообразную, a g- монотонная функция, стремящаяся к нулю при то Н. и.
сходится. Другой признак: если Н. и.
сходится, а функция gмонотонна и ограничена при , то Н. и.
сходится.
Сходимость Н. и. можно выразить в терминах сходящихся рядов: напр., для того чтобы Н. и. (1) сходился, необходимо и достаточно, чтобы для любой последовательности сходился ряд
причем в случае его сходимости сумма ряда совпадает с Н. и. (1).
Понятие Н. и. обобщается для функций многих переменных. Пусть функция f определена на открытом (ограниченном или неограниченном) множестве G n -мерного евклидова пространства и интегрируема по Риману на любом измеримом по Жордану множестве Функцию f наз. интегрируемой в несобственном смысле по множеству G, если для любой последовательности измеримых по Жордану множеств таких, что существует предел
не зависящий от выбора указанной последовательности . Этот предел, если он существует, наз. Н. и.
и, как в одномерном случае, говорят, что этот интеграл сходится. Он существует тогда и только тогда, когда существует интеграл
В этом случае Н. и.
совпадает с интегралом Лебега. Это обстоятельство связано с тем, что при n=1 и данном выше определении Н. и. переход к пределу осуществлялся по весьма специальному классу измеримых по Жордану множеств, а именно по отрезкам. В качестве же были взяты произвольные измеримые по Жордану множества. Впрочем, при сделанное утверждение остается в силе и в том случае, когда в качестве множеств взяты только измеримые по Жордану области. Таким образом, в этом случае понятие Н. и. не приводит к новому понятию по сравнению с интегралом Лебега.
Для Н. и. от функции многих переменных справедлив признак сравнения, аналогичный одномерному случаю. В качестве интегралов сравнения берут
где
Первый сходится при и расходится при , второй сходится при и расходится при .
К Н. и. относятся интегралы в смысле главного значения. Пусть функция f определена на открытом множестве , кроме, быть может, точки , и пусть для любого функция f интегрируема (по Риману или по Лебегу) на множестве есть -окрестность точки х. Тогда если существует предел
то его наз. интегралом в смысле главного значения и обозначают
Если интеграл
существует как Н. и., то он существует и в смысле главного значения. Обратное, вообще говоря, неверно. Напр., Н. и. расходится, а
Аналогично определяют интегралы в смысле главного значения в бесконечно удаленной точке.
Лит.:[1] Ильин В. А., Позняк Э. Г., Основы математического анализа, т. 2, М., 1973; [2] Кудрявцев Л. Д., Курс математического анализа, т. 2, М., 1981; [3] Никольский С. М., Курс математического анализа, 2 изд., т. 2, М., 1975.
Л. Д. Кудрявцев.
Математическая энциклопедия. — М.: Советская энциклопедия.
И. М. Виноградов.
1977—1985.
dic.academic.ru
Несобственный интеграл — это… Что такое Несобственный интеграл?
Определённый интеграл называется несобственным, если выполняется, по крайней мере, одно из следующих условий:
Предел a или b (или оба предела) являются бесконечными;
Функция f(x) имеет одну или несколько точек разрыва внутри отрезка [a, b].
Несобственные интегралы I рода
Пусть определена и непрерывна на множестве от и . Тогда:
Если , то используется обозначение и интеграл называется несобственным интегралом Римана первого рода. В этом случае называется сходящимся.
Если не существует конечного ( или ), то интеграл называется расходящимся к , или просто расходящимся.
Пусть определена и непрерывна на множестве от и . Тогда:
Если , то используется обозначение и интеграл называется несобственным интегралом Римана первого рода. В этом случае называется сходящимся.
Если не существует конечного ( или ), то интеграл называется расходящимся к , или просто расходящимся.
Если функция определена и непрерывна на всей числовой прямой, то может существовать несобственный интеграл данной функции с двумя бесконечными пределами интегрирования, определяющийся формулой:
, где с — произвольное число.
Геометрический смысл несобственного интеграла I рода
Несобственный интеграл выражает площадь бесконечно длинной криволинейной трапеции.
Примеры
Несобственные интегралы II рода
Пусть определена на , терпит бесконечный разрыв в точке x=a и . Тогда:
Если , то используется обозначение и интеграл называется несобственным интегралом Римана второго рода. В этом случае интеграл называется сходящимся.
Если или , то обозначение сохраняется, а называется расходящимся к , или просто расходящимся.
Пусть определена на , терпит бесконечный разрыв при x=b и . Тогда:
Если , то используется обозначение и интеграл называется несобственным интегралом Римана второго рода. В этом случае интеграл называется сходящимся.
Если или , то обозначение сохраняется, а называется расходящимся к , или просто расходящимся.
Если функция терпит разрыв во внутренней точке отрезка , то несобственный интеграл второго рода определяется формулой:
Геометрический смысл несобственных интегралов II рода
Несобственный интеграл выражает площадь бесконечно высокой криволинейной трапеции
Пример
Отдельный случай
Пусть функция определена на всей числовой оси и имеет разрыв в точках .
Тогда можно найти несобственный интеграл
Критерий Коши
1. Пусть определена на множестве от и .
Тогда сходится
2. Пусть определена на и .
Тогда сходится
Абсолютная сходимость
Интеграл называется абсолютно сходящимся, если сходится. Если интеграл сходится абсолютно, то он сходится.
Условная сходимость
Интеграл называется условно сходящимся, если сходится, а расходится.
См. также
Список используемой литературы
Дмитрий Письменный Конспект лекций по высшей математике, часть 1. — Айрис Пресс, 2007. — С. 233-237.
dic.academic.ru
Несобственный интеграл — Википедия. Что такое Несобственный интеграл
Определённый интеграл называется несобственным, если выполняется, по крайней мере, одно из следующих условий.
Область интегрирования является бесконечной. Например, является бесконечным промежутком [a,+∞){\displaystyle [a,+\infty )}.
Функция f(x){\displaystyle f(x)} является неограниченной в окрестности некоторых точек области интегрирования.
Если интервал [a,b]{\displaystyle [a,b]} конечный и функция интегрируема по Риману, то значение несобственного интеграла совпадает со значением определённого интеграла.
Несобственные интегралы I рода
Несобственный интеграл первого рода
Пусть f(x){\displaystyle f(x)} определена и непрерывна на интервале [a,+∞){\displaystyle [a,+\infty )} и ∀A>a ∃∫aAf(x)dx{\displaystyle \forall A>a\ \exists \int \limits _{a}^{A}f(x)dx}. Тогда:
Если ∃limA→+∞∫aAf(x)dx=I∈R{\displaystyle \exists \lim _{A\to +\infty }\int \limits _{a}^{A}f(x)dx=I\in \mathbb {R} }, то используется обозначение I=∫a+∞f(x)dx{\displaystyle I=\int \limits _{a}^{+\infty }f(x)dx} и интеграл называется несобственным интегралом Римана первого рода. В этом случае I=∫a+∞f(x)dx{\displaystyle I=\int \limits _{a}^{+\infty }f(x)dx} называется сходящимся.
Если не существует конечного limA→+∞∫aAf(x)dx{\displaystyle \lim _{A\to +\infty }\int \limits _{a}^{A}f(x)dx} (±∞{\displaystyle \pm \infty } или ∄{\displaystyle \nexists }), то интеграл ∫a+∞f(x)dx{\displaystyle \int \limits _{a}^{+\infty }f(x)dx} называется расходящимся к «∞{\displaystyle \infty }», «±∞{\displaystyle \pm \infty }», или просто расходящимся.
Пусть f(x){\displaystyle f(x)} определена и непрерывна на множестве от (−∞,b]{\displaystyle (-\infty ,b]} и ∀B<b⇒∃∫Bbf(x)dx{\displaystyle \forall B<b\Rightarrow \exists \int \limits _{B}^{b}f(x)dx}. Тогда:
Если ∃limB→−∞∫Bbf(x)dx=I∈R{\displaystyle \exists \lim _{B\to -\infty }\int \limits _{B}^{b}f(x)dx=I\in \mathbb {R} }, то используется обозначение I=∫−∞bf(x)dx{\displaystyle I=\int \limits _{-\infty }^{b}f(x)dx} и интеграл называется несобственным интегралом Римана первого рода. В этом случае I=∫−∞bf(x)dx{\displaystyle I=\int \limits _{-\infty }^{b}f(x)dx} называется сходящимся.
Если не существует конечного limB→−∞∫Bbf(x)dx{\displaystyle \lim _{B\to -\infty }\int \limits _{B}^{b}f(x)dx} (±∞{\displaystyle \pm \infty } или ∄{\displaystyle \nexists }), то интеграл ∫−∞bf(x)dx{\displaystyle \int \limits _{-\infty }^{b}f(x)dx} называется расходящимся к «∞{\displaystyle \infty }», «±∞{\displaystyle \pm \infty }», или просто расходящимся.
Если функция f(x){\displaystyle f(x)} определена и непрерывна на всей числовой прямой, то может существовать несобственный интеграл данной функции с двумя бесконечными пределами интегрирования, определяющийся формулой:
∫−∞+∞f(x)dx=∫−∞cf(x)dx+∫c+∞f(x)dx{\displaystyle \int \limits _{-\infty }^{+\infty }f(x)dx=\int \limits _{-\infty }^{c}f(x)dx+\int \limits _{c}^{+\infty }f(x)dx}, где с — произвольное число.
Геометрический смысл несобственного интеграла I рода
Несобственный интеграл первого рода выражает площадь бесконечно длинной криволинейной трапеции.
Пусть f(x){\displaystyle f(x)} определена на (a,b]{\displaystyle (a,b]}, терпит бесконечный разрыв в точке x = a и ∀δ>0⇒∃∫a+δbf(x)dx=I(δ){\displaystyle \forall \delta >0\Rightarrow \exists \int \limits _{a+\delta }^{b}f(x)dx={\mathcal {I}}(\delta )}. Тогда:
Если ∃limδ→0+0I(δ)=I∈R{\displaystyle \exists \lim _{\delta \to 0+0}{\mathcal {I}}(\delta )=I\in \mathbb {R} }, то используется обозначение I=∫abf(x)dx{\displaystyle I=\int \limits _{a}^{b}f(x)dx} и интеграл называется несобственным интегралом Римана второго рода. В этом случае интеграл называется сходящимся.
Если limδ→0+0I(δ)=∞(±∞{\displaystyle \lim _{\delta \to 0+0}{\mathcal {I}}(\delta )=\infty \;(\pm \infty } или ∄){\displaystyle \nexists )}, то обозначение сохраняется, а I=∫abf(x)dx{\displaystyle {\mathcal {I}}=\int \limits _{a}^{b}f(x)dx} называется расходящимся к «∞{\displaystyle \infty }», «±∞{\displaystyle \pm \infty }», или просто расходящимся.
Пусть f(x){\displaystyle f(x)} определена на [a,b){\displaystyle [a,b)} , терпит бесконечный разрыв при x = b и ∀δ>0⇒∃∫ab−δf(x)dx=I(δ){\displaystyle \forall \delta >0\Rightarrow \exists \int \limits _{a}^{b-\delta }f(x)dx={\mathcal {I}}(\delta )}. Тогда:
Если ∃limδ→0+0I(δ)=I∈R{\displaystyle \exists \lim _{\delta \to 0+0}{\mathcal {I}}(\delta )=I\in \mathbb {R} }, то используется обозначение I=∫abf(x)dx{\displaystyle I=\int \limits _{a}^{b}f(x)dx} и интеграл называется несобственным интегралом Римана второго рода. В этом случае интеграл называется сходящимся.
Если limδ→0+0I(δ)=∞(±∞{\displaystyle \lim _{\delta \to 0+0}{\mathcal {I}}(\delta )=\infty \;(\pm \infty } или ∄){\displaystyle \nexists )}, то обозначение сохраняется, а I=∫abf(x)dx{\displaystyle {\mathcal {I}}=\int \limits _{a}^{b}f(x)dx} называется расходящимся к «∞{\displaystyle \infty }», «±∞{\displaystyle \pm \infty }», или просто расходящимся.
Если функция f(x){\displaystyle f(x)} терпит разрыв во внутренней точке c{\displaystyle c} отрезка [a;b]{\displaystyle [a;b]}, то несобственный интеграл второго рода определяется формулой:
Дмитрий Письменный. Конспект лекций по высшей математике, часть 1. — Айрис Пресс, 2007. — С. 233-237.
wiki.sc
Несобственные интегралы — это… Что такое Несобственные интегралы?
обобщение классического понятия интеграла на случай неограниченных функций и функций, заданных на бесконечном промежутке интегрирования (см. Интеграл). Определённый интеграл как предел интегральных сумм Римана может существовать (иметь определённое конечное значение) лишь для ограниченных функций, заданных на конечном интервале. Поэтому, если интервал интегрирования или подынтегральная функция не ограничены, для определения интеграла требуется ещё один предельный переход: получающиеся при этом интегралы называются несобственными интегралами.
Если функция f (x) интегрируема на любом конечном отрезке [a, N] и если существует
то его называют Н. п. функции f (x) на интервале [а, ∞] и обозначают
В этом случае говорят, что Н. и. сходится. Когда этот предел, а значит и Н. и., не существует, то иногда говорят, что Н. и. расходится. Например,
сходится при γ > 1 и расходится при γ ≤ 1. Аналогично определяют Н. и. на интервалах
[—∞, b] и [—∞, ∞].
Если функция f (x), заданная на отрезке [a, b], не ограничена в окрестности точки a, но интегрируема на любом отрезке [а + ε, b], 0 b — a и если существует
то его называют Н. и. функции f (x) на [а, b] и записывают обычным образом:
Аналогично поступают, если f (x) не ограничена в окрестности точки b.
Если существует Н. и.
или
то говорят, что Н. и.
или
абсолютно сходится: если же последние интегралы сходятся (но первые расходятся), то Н. и.
или
называются условно сходящимися.
Задачи, приводящие к Н. и., рассматривались в геометрической форме Э. Торричелли и П. Ферма в 1644. Точные определения Н. и. даны О. Коши в 1823. Различие условно и абсолютно сходящихся Н. и. установлено Дж. Стоксоми П. Г. Л. Дирихле (1854). Ряд работ математиков 19 в. посвящен вычислению Н. и. в случаях, когда соответствующая первообразная не выражается через элементарные функции. Основными приемами вычисления Н. и. являются дифференцирование и интегрирование по параметру, разложение в ряды, применение теории вычетов. Значения многих Н. и. приводятся в различных таблицах.
Н. и. имеют важное значение во многих областях математического анализа и его приложений. В теории специальных функций (цилиндрических функций, ортогональных многочленов и др.) одним из основных способов изучения является изображение функций в виде Н. и., зависящих от параметра, например
(см. Гамма-функция). К Н. и. относится и Фурье интеграл, а также интегралы, встречающиеся при др. интегральных преобразованиях. Решения краевых задач (См. Краевые задачи)математической физики записываются кратными Н. и. с неограниченной подинтегральной функцией. В теории вероятностей важное значение имеет Н. и.
в теории диффракции света — Н. и.
В ряде случаев расходящимся Н. и. можно приписать определённое значение (см. Суммирование). В частности, если интеграл
расходится, но существует
то А называется главным значением Н. и. и обозначают
Так,
Аналогично вводится главное значение Н. и. от неограниченных функций. В работах Н. И. Мусхелишвили и его учеников построена теория интегральных уравнений, содержащих Н. и., понимаемые в смысле главного значения.
Лит.: Смирнов В. И., Курс высшей математики, 20 изд., т. 2, М. — Л., 1967; Фихтенгольц Г. М., Курс дифференциального и интегрального исчисления, 7 изд. т. 2, М., 1969; Кудрявцев Л. Д., Математический анализ, т. 1, М., 1970.
partners.academic.ru
Несобственные интегралы
Пример. Сходится ли интеграл ∫1
dx
?
sin x
0
Решение:
Функция f(x) = sin1 x имеет на [0; 1] единственный разрыв в точке х = 0. Рассмотрим
функцию ϕ(х) = 1 . Интеграл x
1
dx
= lim
1
dx
= limln x 1 = 0 − limlnε
0
x
ε →0
0+ε
x
ε →0
|ε
ε →0
∫
∫
расходится. И так как
lim
f (x)
= lim
x
= 1,
x→0
ϕ (x)
x→0 sin x
то интеграл ∫1
dx
также расходится.
0 sin x
Геометрические приложения
Схемы применения определенного интеграла
Пусть требуется найти значение какой-либо геометрической или физической величины А (площадь фигуры, объем тела, давление жидкости на вертикальную пластину и т.д.), связанной с отрезком [а; b] изменения независимой переменной х. Предполагается, что эта величина А аддитивна, т. е. такая, что при разбиении отрезка [а; b] точкой с (а; b) на части [а, с] и [с; b] значение величины А, соответствующее всему отрезку [a; b], равно сумме ее значений, соответствующих [а; с] и [с, b].
Для нахождения этой величины А можно руководствоваться одним из двух методов: методом интегральных сумм или методом дифференциала.
Первый метод базируется на определении определенного интеграла.
1 Точками x0 = a, x1, … xn = b разбить отрезок [а; b] на n частей. В соответствии с этим, интересующая нас величина А разобьется на n «элементарных слагаемых» Ai (i = 1, …, n): A = A1 + A2 + … + An.
2. Представить каждое «элементарное слагаемое» в виде произведения некоторой функции (определяемой из условия задачи), вычисленной в произвольной точке соответствующего отрезка на его длину: Ai ≈ f(ci) xi.
При нахождении приближенного значения Ai допустимы некоторые упрощения: дугу на малом участке можно заменить хордой, стягивающей ее концы; переменную скорость на малом участке можно приближенно считать постоянной и т.д.
Получим приближенное значение величины А в виде интегральной суммы:
n
A ≈ f(c1) x1 + … + f(cn) xn = ∑ f (ci ) xi .
i=1
3.Искомая величина А равна пределу интегральной суммы, т.е.
n
b
A = limn→∞
∑ f (ci )
xi = ∫ f (x)dx.
i=1
a
Указанный «метод сумм», как видим, основан на представлении интеграла как о сумме бесконечно большого числа бесконечно малых слагаемых.
studfile.net
2.1. Несобственные интегралы для «чайников»
Это «родственник» определённого интеграла. …Нормальное такое определение :). И сразу возникает вопрос: чем отличается несобственный интеграл от «собрата»? Он может отличаться пределами интегрирования: – то есть, один или даже оба предела бесконечны, при этом подынтегральная функция непрерывна на промежутке интегрирования.
Такие интегралы получили название несобственные интегралы первого рода.
Кроме того, несобственный интеграл может быть «внешне похож» на определённый интеграл и иметь вид . Но есть один нюанс. Подынтегральная функция не определена в точке или . Или на обоих концах. Или даже во внутренних точках отрезка .
Это так называемые несобственные интегралы второго рода.
Что значит решить несобственный интеграл? В отличие от определённого интеграла, тут есть три варианта. Решить несобственный интеграл – это значит найти конечное число, либо получить бесконечность, либо выяснить, что несобственного интеграла не существует.
1) Если несобственный интеграл равен конечному числу, то говорят, что он сходится. Число может быть как положительным, так и отрицательным. Или нулём.
2) Если несобственный интеграл равен бесконечности (со знаком «плюс» или «минус»), то говорят, он расходится.
3) И в ряде случаев несобственного интеграла может вовсе не существовать. Даже если подынтегральная функция непрерывна на промежутке интегрирования! (вспоминаем, что определённый интеграл при этом условии существует всегда).
Как решить несобственный интеграл? С помощью той же формулы Ньютона-Лейбница. С некоторыми особенностями.
И здесь вы должны понимать и уметь решать несложные пределы функций.
В чём смысл несобственного интеграла? Геометрически – это тоже площадь (если интеграл существует). Но площадь своеобразная. И с этим своеобразием мы познакомимся прямо на следующей странице:
2.2. Несобственный интеграл первого рода
1.11. А если подынтегральная функция нечётная?
| Оглавление |
Полную и свежую версию данного курса в pdf-формате, а также курсы по другим темам можно найти здесь.
Также вы можете изучить эту тему подробнее – просто, доступно, весело и бесплатно!
С наилучшими пожеланиями, Александр Емелин
mathprofi.com
§1. Несобственные интегралы 1-го рода
– 68–
Тема НЕСОБСТВЕННЫЕ ИНТЕГРАЛЫ
В
теме «Определенный интеграл» было
рассмотрено понятие определенного
интеграла для случая конечного промежуткаи ограниченной функции(см. теорему 1 из §3). Теперь займемся
обобщением этого понятия для случаев
бесконечного промежутка и неограниченной
функции. Необходимость такого обобщения
показывают, например, такие ситуации.
1.
Если, используя формулу для длины дуги,
попытаться вычислить длину четверти
окружности ,,
то придем к интегралу от неограниченной
функции:
,
где .
2.
Пусть тело массой движется
по инерции в среде с силой сопротивления ,
где— скорость тела. Используя второй закон
Ньютона (,
гдеускорение),
получим уравнение:,
где.
Нетрудно показать, что решением этого
(дифференциального!) уравнения является
функцияЕсли
нам потребуется вычислить путь, пройденный
телом до полной остановки, т.е. до момента,
когда ,
то придем к интегралу по бесконечному
промежутку:
I Определение
Пусть
функция определена и непрерывна на промежутке.
Тогда для любогоона интегрируема на промежутке,
то есть существует интеграл.
Определение
1.
Конечный или бесконечный предел этого
интеграла при называют несобственным интегралом 1-го
рода от функциипо промежуткуи обозначают символом.
При этом, если указанный предел конечен,
то несобственный интеграл называют
сходящимся, в противном случае (или не существует ) – расходящимся.
Итак,
по определению
(1)
Примеры
1..
2..
3.– не существует.
Несобственный
интеграл из примера 1 сходится, в примерах
2 и 3 интегралы расходятся.
II Формула Ньютона – Лейбница для несобственного интеграла первого рода
Пусть — некоторая первообразная для функции(сущест-вует на,
т.к.— непрерывна). Тогда
Отсюда
ясно, что сходимость несобственного
интеграла (1) равносильна существованию
конечного предела.
Если этот предел обозначить,
то можно написать для интеграла (1)
формулу Ньютона-Лейбница:
,
где .
Примеры.
4. .
5. .
6.
Более сложный пример: .
Сначала найдем первообразную:
Теперь
можем найти интеграл , учитывая,
что:
.
III Свойства
Приведем
ряд свойств несобственного интеграла
(1), которые вытекают из общих свойств
пределов и определенного интеграла:
интегралы исходятся или расходятся одновременно;
если ,
то интегралыисходятся или рас-ходятся одновременно;
если
интеграл сходится, то.
IV Другие определения
Определение
2.
Если непрерывна
на ,
то
.
Определение
3.
Если непрерывна
на,
то принимают по определению
(–
произвольное),
причем
несобственный интеграл в левой части
сходится, если только оба ин-теграла в
правой части сходятся.
Для
этих интегралов, как и для интеграла
(1) можно написать соответствующие
формулы Ньютона – Лейбница.
Пример
7.
§2.
Признаки сходимости несобственного
интеграла 1-го рода
Чаще
всего несобственный интеграл вычислить
по определению не-возможно, поэтому
используют приближенное равенство
(для
больших ).
Однако,
это соотношение имеет смысл лишь для
сходящихся интегралов. Необходимо иметь
методы выяснения поведения интеграла
минуя определение.
I Интегралы от положительных функций
Пусть на . Тогда определенный интеграл как функция верхнего предела есть
функция возрастаю-щая (это следует из
общих свойств определенного интеграла).
Теорема
1.
Несобственный интеграл 1го рода от неотрицательной функ-ции сходится
тогда и только тогда, когда функция остается
ограниченной при увеличении.
Эта
теорема – следствие общих свойств
монотонных функций. Практического
смысла теорема почти не имеет, но
позволяет получить т.н. признаки
сходимости.
Доказательство.
Обозначим: и.
Так как,
то.
Пусть интегралсходится, тогда (в силу теоремы 1) функция‒ ограничена. Но тогда иограничена,
а значит, интегралтоже сходится. Аналогично доказывается
и вторая часть теоремы.
Этот
признак не применим в случае расходимости
интеграла от или сходимости интеграла от.
Этот недостаток отсутствует у 2-го
признака сравнения.
Теорема
3 (2-й признак сравнения). Пусть функции инепрерывны и неотрицательны на.
Тогда, еслипри,
то несобственные интегралыисходятся или расходятся одновременно.
Доказательство.
Из условия теоремы получим такую цепочку
равно-сильных утверждений:
, ,
.
Пусть,
например, .
Тогда:
.
Применим
теорему 2 и свойство 1) из §1 и получим
утверждение теоремы 3.
В
качестве эталонной функции, с которой
сравнивают данную, высту-пает степенная
функция ,.
Предлагаем студентам самим доказать,
что интеграл
сходится
при и расходится при.
Примеры.
1. .
Рассмотрим
подынтегральную функцию на промежутке :
, .
Интеграл сходится, ибо.
По 2-му признаку сравнения сходится и
интеграл,
а в силу свойства 2) из §1 сходится и
исход-ный интеграл.
2..
Так
как ,
тоcуществует такое, что при.
Для таких значений переменной:
.
Известно,
что логарифмическая функция растет
медленнее степенной, т.е.
,
а
значит, начиная с некоторого значения
переменной, эта дробь меньше 1. Поэтому
.
Интеграл сходится как эталонный. В силу 1-го
признака сравнения сходится и.
Применяя 2-й признак, получим, что и
интегралсходится. И снова свойство 2) из §1
доказывает сходимость исходного
интеграла.
dv = dl/dt, т.е. путевая скорость вдоль рассматриваемой траектории
(6)
Нормальное ускорение
(7)
Скорость свободного падения тела
(8)
Время тела при свободном падении
(9)
Время при равномерном движении по окружности
(10)
Скорость равномерного движения по окружности
(11)
Угловая (мгновенная) скорость равномерного движения по окружности
Единица измерения угловой скорости — радианы в секунду
(12)
Скорость равноускоренного движения по окружности
(13)
Угловая (мгновенная) скорость равноускоренного движения по окружности
— версия для печати
Определение
Кинематикой называется раздел физики, занимающийся исследованием законов движения идеальных тел
Пояснение
Под чертой вверху буквы подразумевается знак вектора.
Если у вас есть мысли или идеи по поводу данной таблицы или, например, вы считаете, что полезно было бы создать определенную
вспомогательную памятку, то мы обязательно рассмотрим ваше предложение, которое можно изложить по ссылке (где вы также можете поделиться с нами любыми мыслями по поводу сайта scolaire. ru).
Мы готовы устранить любые неудобства, связанные с использованием данной таблицы, или ей подобных, которые можно найти в разделе
«Физика».
Формулы по кинематике, динамике, законам сохранения, молекулярной физике, электричеству, магнетизму, оптике. Тест
Формулы по кинематике, динамике, законам сохранения, молекулярной физике, электричеству, магнетизму, оптике. Тест — курсы по физике
Skip navigation
Элементы математики
действия с векторами
выражение неизвестной
Физические величины
Единицы измерения
внесистемные единицы
Постоянные величины в физике
плотность вещества
предел прочности, модуль Юнга
скорость звука
удельная теплота
диэлектрическая проницаемость
удельное сопротивление
электрохимический эквивалент
Формулы
I. Механика
Кинематика
равномерное движение
относительность движения
неравномерное движение
равноускоренное движение
ускорение свободного падения
графики движения
движение по окружности
параболическое движение
Динамика
закон тяготения
законы Ньютона
силы в природе
равнодействующая сила
Законы сохранения
импульс тела, импульс силы
закон сохранения импульса
работа и мощность
кинетическая и потенциальная энергии
закон сохранения энергии
Статика
плечо и момент силы
условия равновесия
центр тяжести, центр масс
Колебания и волны
колебательное движение
гармонические колебания
маятники
превращение энергии при колебаниях
упругие волны
звуковые волны
II. Молекулярная физика
Молекулярная физика
основные положения мкт
давление
основное уравнение мкт, температура
уравнение идеального газа
изопроцессы
свойства жидкостей*
свойства твердых тел
Термодинамика
количество теплоты
работа, внутренняя энергия
первый закон термодинамики
второй закон термодинамики
тепловые двигатели
III. Основы электродинамики
Электричество
электрический заряд
закон Кулона
напряженность поля
потенциал и работа поля
диэлектрики, проводники
электроемкость, конденсаторы
энергия конденсатора
Электрический ток
электрический ток, сила и плотность
закон Ома для участка цепи
работа и мощность тока
закон Ома для замкнутой цепи
электрический ток в различных средах
электрические явления
Магнетизм
магнитное поле
сила Ампера
сила Лоренца
Электромагнетизм
магнитный поток
закон электромагнитной индукции
самоиндукция, энергия поля
электромагнитные колебания
электромагнитные волны
переменный ток
трансформатор*
IV. Оптика
Волновая оптика
свет как электромагнитные волны
интерференция
дифракция
Геометрическая оптика
законы распространения света
линзы, оптические приборы
V. Теория относительности
Теория относительности
постулаты теории относительности
VI. Квантовая физика
Световые кванты
фотон
фотоэффект
квантовые постулаты Бора
излучение и поглощение света
Атомное ядро
энергия связи ядра
ядерные реакции
закон радиоактивного распада
элементарные частицы и их свойства
Современная физика*
физика элементарных частиц
мир внутри атомного ядра
время расщепляем на мгновения
нанотехнологии и нанофизика
вещество в экстремальных состояниях
Закрыть
формулы, определения, методы решения задач
Кинематика — это специальный раздел теоретической механики. Направление сформировалось несколько позднее, чем статика и динамика: во второй половине XIX столетия. Первые исследования в области кинематики были посвящены огнестрельному оружию. Ученые стремились понять процесс полета снаряда, производили расчет траектории его движения. В дальнейшем кинематика как научное направление получило широкое распространение и существенно повлияло на развитие технического прогресса.
Кинематика — описание
Кинематика является разделом механики, цель которого — изучение механического движения тел с пренебрежением к причинам, вызывающим это движение.
Механика представляет собой научную область физики, которой посвящены исследования механического движения тел. Основной целью данного направления служит определение точного положения тела в пространстве в любой момент времени. Важным понятием этого раздела является материальная точка в виде тела с определенной массой и размерами, которыми можно пренебречь для решения задачи при наличии следующих условий:
Путь, который преодолевает тело, существенно больше, чем его размеры.
Расстояние между телами значительно превышает их размеры.
Объект совершает поступательное движение.
Движение тела рассматривают в системе отсчета, состоящей из системы координат и прибора, измеряющего время. Траекторией называют линию, которую объект описывает, совершая движение. Путь является скалярной величиной, определяемой как длина траектории. Перемещением обозначают вектор, который соединяет начальное и конечное положение тела, преодолеваемое им в течение определенного промежутка времени.
Совершая движение, тело может только увеличивать пройденный путь, при этом перемещение увеличивается или уменьшается. К примеру, уменьшение перемещения наблюдается во время обратного движения тела. Если объект движется прямолинейно в одном направлении, то путь определяется модулем перемещения. В случае криволинейного движения — путь превышает перемещение. При рассмотрении замкнутой траектории перемещение будет равно нулю.
Теория и формулы
Благодаря многолетним исследованиям в области кинематики ученым удалось вывести определенные закономерности движения тела. С помощью справедливых уравнений представляется возможным ответить на многие вопросы о разных характеристиках, которые изменяются либо остаются постоянными во время движения объектов.
Путь, время, скорость
Расстояние представляет собой удаленность одной точки положения тела от другой. Тело преодолевает путь, который представляет собой важную характеристику механического движения. Общепринятым обозначением пути является латинская буква s. Данный параметр измеряют метрами и километрами, если речь идет о больших расстояниях.
Скорость представляет собой путь, который тело преодолело в течение единицы времени. В качестве единицы времени часто используют 1 час, 1 минуту, 1 секунду. Для расчета скорости необходимо определить отношение пути к времени движения. В случае, когда в условиях задачи расстояние измеряется в метрах, а время пути — в секундах, то скорость следует рассчитывать в метрах в секунду (м/с). Для обозначения скорости используют латинскую букву \(v\).
Нередко требуется определить время пути. Данный параметр обозначают с помощью латинской буквы \(t\).
Важно отметить, что скорость, путь и время взаимосвязаны. При известных характеристиках скорости и времени можно определить расстояние, которое преодолело тело. Путь в данном случае равен произведению скорости и времени, рассчитывается по формуле:
\(s=v\times t\)
При известных величинах времени и расстояния достаточно просто определить скорость движения тела, руководствуясь следующим уравнением:
\(v=\frac{s}{t}\)
Равномерное движение
Равномерным движением называют движение тела, которое совершает равные перемещения в течение любых равных промежутков времени.
Источник: goodfon.ru
Скорость при равномерном движении определяется как отношение перемещения ко времени, в течение которого данное перемещение было совершено. Уравнение имеет следующий вид:
\(\vec{v}=\frac{\vec{s}}{t}\)
\(\vec{v}=const\)
Проекция вектора скорости на ось ОХ выглядит таким образом:
\(v_{x}=\frac{s_{x}}{t}\)
\(v_{x}=const\)
Если вектор скорости спроецировать на ось координат, то она будет равна быстроте изменения данной координаты:
\(v_{x}=\frac{x-x_{0}}{t}\)
Прямолинейное равноускоренное движение
Прямолинейным равноускоренным движением называют движение по прямой траектории, для которого характерно постоянное ускорение.
Ускорение для прямолинейного равноускоренного движения обозначают следующим образом:
\(\vec{a}=const\)
При таком движении можно наблюдать увеличение или уменьшение скорости. Чтобы определить скорость, необходимо выполнить следующий расчет:
\(\vec{v}=\vec{v}_{0}+\vec{a}t\)
Если тело разгоняется в проекции оси ОХ, то скорость можно определить по формуле:
\(v_{x}=v_{0x}+a_{x}t\)
a>0, движение является равноускоренным.
Источник: fizi4ka.ru
Во время торможения в проекции на ось ОХ скорость рассчитывают следующим образом:
\(v_{x}=v_{0x}-a_{x}t\)
а<0, движение является равнозамедленным.
Источник: fizi4ka.ru
Графически зависимость ускорения от времени, то есть график ускорения во время равноускоренного движения тела, можно представить в виде:
Источник: fizi4ka.ru
График ускорения, характеризующий равноускоренное движение тела, представляет собой прямую, которая параллельна оси времени:
график 1 находится над осью t, тело совершает разгон, ах>0;
график 2 размещен под осью t, тело тормозит, ах<0.
Графически скорость или проекция скорости изображается в виде зависимости скорости от времени:
Источник: fizi4ka. {2}}\)
Дальность полета тела соответствует уравнению:
\(l=v_{0x}t=v_{0x}\sqrt{\frac{2h_{0}}{g}}\)
Вычислить угол между вектором скорости и осью ОХ можно с помощью формулы:
Рассмотрим путь велосипедиста из одного населенного пункта в другой. Половина расстояния была преодолена со скоростью 12 км/ч (\(v_1\)). Далее половину оставшегося времени он ехал со скоростью 6 км/ч (\(v_2\)). Остаток расстояния путник преодолел пешком со скоростью 4км/ч (\(v_3\)). Необходимо рассчитать среднюю скорость на всем пути следования велосипедиста.
Решение
Данный пример относится к теме равномерного прямолинейного движения одного тела. Процесс можно изобразить схематично:
Источник: pandia.ru
\(S = S_1 + S_2 + S_3\)
\(t = t_1 + t_2 + t_3\)
На каждый отрезок пути необходимо составить уравнение движения:
\(S_1 = v_1t_1\)
\(S_2 = v_2t_2\)
\(S_3 = v_3t_3\)
Далее можно представить дополнительные условия задачи:
Тело подбросили вертикально вверх. Начальная скорость при этом составила 3,13 м/с (\(v_0\)). В момент, когда данное тело достигло максимальную высоту полета, из начального пункта подбросили второе тело с такой же начальной скоростью, как у первого. Необходимо определить на каком расстоянии от точки бросания встретятся тела. {2}}=\frac{9,81}{0,17}=57,7\)
Ответ: камень упал с высоты \(57,7\) м.
Решение задач по кинематике основано на простых формулах. Успешность результата зависит от умения грамотно применять справедливые уравнения в том или ином случае. Бывают ситуации, когда в процессе изучения физики возникают некоторые трудности. Простым решением будет обратиться к порталу Феникс.Хелп.
Осн. формулы и метод. рекомендации по решению задач на кинематику МТ
Ближайшие темы будут посвящены решению
задач на движение тел, без учета причин, вызвавших это движения, т.е. решению
задач по кинематике.
Но для того, чтобы начать рассмотрение решений задач по данной
теме, необходимо вспомнить основные формулы, связанные с этим разделом. Для удобства,
сведём все формулы в таблицу.
Основные формулы равномерного прямолинейного движения
Формула
Описание
формулы
Перемещение
тела за промежуток времени t, где –
скорость тела, sx, vx – проекции перемещения и скорость на ось Ох.
Путь
за промежуток времени t.
Закон
сложения скоростей в классической механике.
Кинематическое
уравнение равномерного движения, где х — координата тела в момент
времени t, х0 — начальная
координата тела.
Основные формулы равноускоренного прямолинейного движения
Формула
Описание
формулы
Скорость
тела в момент времени t, где –
ускорение тела, –
скорость тела в начальный момент времени.
Перемещение
тела за промежуток времени t.
Кинематическое
уравнение равноускоренного движения.
Основные формулы движения тела по окружности с постоянной
по модулю скоростью.
Формула
Описание
формулы
Линейная
скорость тела, где l — длина дуги окружности,
пройденной телом за промежуток времени Δt.
Угловая
скорость тела, где Dj –
угол поворота радиус-вектора движущегося по окружности тела за промежуток
времени Dt.
Связь
линейной скорости с угловой, где R — радиус
окружности.
Период
вращения, где N — число оборотов тела за промежуток
времени Δt.
Частота
вращения.
Связь
между линейной скоростью, периодом вращения и частотой.
Центростремительное
ускорение.
Известно, что для большей наглядности движение можно
описывать с помощью графиков.
Давайте рассмотрим в сравнении графики для равномерного и
равноускоренного движения.
Известно, что при равномерном движении скорость тела не
изменяется с течением времени. Поэтому графиком скорости, в этом случае, будет
прямая линия, параллельная оси времени. При равноускоренном движении тела,
неизменной величиной является ускорение. Поэтому графиком ускорения будет
являться также прямая линия, параллельная оси времени.
По графику скорости для равномерного движения, можно
определить путь, пройденный телом за некоторый промежуток времени. Для этого
достаточно определить площадь прямоугольника, образованного графиком скорости и
осью времени.
Известно, что перемещение тела при равномерном движении
линейно зависит от времени, поэтому графиком перемещения является прямая линия
вида
y = kx.
Наклон же графика к оси времени зависит от модуля скорости. При
равноускоренном движении линейно зависимой величиной является скорость тела.
Поэтому графиком скорости является прямая линия вида
y = kx +b.
Используя график скорости для равноускоренного движения можно
определить перемещение тела за некоторый промежуток времени. Для этого
необходимо определить площадь прямоугольной трапеции или прямоугольного
треугольника, ограниченных графиком скорости и осью времени.
График зависимости координаты от времени при равномерном
движении, то есть график движения, представлен на рисунке ниже. По этому
графику можно определить: координату тела в любой момент времени, путь,
пройденный телом за некоторый промежуток времени, кратчайшее расстояние между
телами в любой момент времени, а также момент и место встречи тел.
А графиком перемещения при равноускоренном движении
является парабола, положение вершины которой зависит от направлений
начальной скорости и ускорения. Так, если проекция ускорения отрицательна,
то возможны следующие три вида графика перемещения:
– когда проекция начальной скороститела равна нулю;
– когда проекция начальной скорости тела меньше нуля;
– когда проекция начальной скорости тела больше нуля.
Если проекция ускорения положительна, то здесь также возможны
три случая:
– когда начальная скорость тела равна нулю;
– когда проекция начальной скорости больше нуля;
– когда проекция начальной скорости меньше нуля.
Методические рекомендации по решению задач на кинематику
материальной точки
1) Сделать схематический рисунок, который лучше всего
представить в виде траектории движущейся точки с изображением векторов
перемещения, скорости и ускорения.
2) Выбрать систему отсчета (то есть тело отсчета, связанную с
ним систему координат и начало отсчета времени) на основании тщательного
анализа условия задачи. Рациональный выбор системы отсчета, как правило,
значительно упрощает решение задачи. При выборе положительных направлений
координатных осей необходимо руководствоваться направлением движения (то есть
направлением вектора скорости) или направлением вектора ускорения.
3) Составить на основании законов движения систему уравнений
в векторном виде для всех тел, участвующих в движении. А затем в скалярной
форме, спроецировав на координатные оси эти векторные уравнения движения. При
записи этих уравнений не забыть привести в соответствие знаки проекций скорости
и ускорения с направлением координатных осей. При необходимости дополнить
систему уравнений соотношениями, составленными на основе данных задачи и
конкретной ситуации, описанной в ней.
4) Решить полученную систему уравнений относительно искомых
величин в общем виде, убедиться в соответствии единиц измерения и проделать
числовые расчеты.
Следование этим простым рекомендациям позволит вам успешнее
справляться с решением задач на кинематику материальной точки.
Законы кинематики формулы. Кинематика основные понятия, законы и формулы. Свободное падение по вертикали
Для того чтобы понять, что изучает механика, необходимо рассмотреть, что означает движение в самом общем смысле. Значение этого слова подразумевает под собой изменение чего-либо. Например, политическое движение выступает за равноправие разных слоев населения вне зависимости от их расовой принадлежности. Раньше его не было, затем что-то изменилось и теперь каждый человек имеет равные права. Это движение цивилизации вперед. Еще пример — экологическое. В прошлом, выбравшись на природу, никто не задумывался о том, что оставляет после себя мусор. Сегодня же любой цивилизованный человек соберет его за собой и отвезет в специально отведенное место для дальнейшей утилизации.
Что-то подобное можно наблюдать и в механике. При механическом движении изменяется положение тела в пространстве относительно других предметов с течением времени. Основная задача механики — указать, где находится объект в любой момент, учитывая даже тот, который еще не наступил. То есть, предсказать положение тела в заданное время, а не только узнать, где именно в пространстве оно находилось в прошлом.
Кинематика — это раздел механики, который изучает движение тела, не анализируя его причины. Это значит, что она учит не объяснять, а описывать. То есть, придумать способ, с помощью которого можно было бы задать положение тела в любой момент времени. Основные понятия кинематики включают в себя скорость, ускорение, расстояние, время и перемещение.
Сложность в описании движения
Первая проблема, с которой сталкивается кинематика — это то, что у каждого тела есть определенный размер. Допустим, необходимо описать движение какого-нибудь предмета. Это значит научиться обозначать его положение в любой момент времени. Но каждый предмет занимает в пространстве какое-то место. То есть, что все части этого объекта в один и тот же момент времени занимают разное положение.
Какую точку в таком случае необходимо взять для описания нахождения всего предмета? Если учитывать каждую, то расчеты окажутся слишком сложными. Поэтому решение ответа на этот вопрос можно максимально упростить. Если все точки одного тела движутся в одинаковом направлении, то для описания движения достаточно одной такой, которую содержит это тело.
Виды движения в кинематике
Существует три типа:
Поступательным называется движение, при котором любая прямая проведенная в теле остается параллельной самой себе. Например, автомобиль, который движется по шоссе, совершает такой вид движения.
Вращательным называется такое движение тела при котором все его точки движутся по окружностям с центрами, лежащими на одной прямой, называемой осью вращения. Например, вращение Земли относительно своей оси.
Колебательным называется движение, при котором тело повторяет свою траекторию через определенный отрезок времени. Например, движение маятника.
Основные понятия кинематики — материальная точка
Любое сложное движение можно описать как комбинацию двух простейших видов — поступательного и вращательного. Например колесо автомобиля или юла, стоящая на движущейся прямо платформе, участвуют одновременно в этих двух типах перемещения.
Но что делать, если движение тела нельзя представить в виде комбинации? Например, если автомобиль едет по ухабистой дороге, его положение будет меняться очень сложным образом. Если рассчитывать только то, что этот транспорт перемещается из одного города в другой, то в такой ситуации становится не важно какого размера тело движется из точки А в точку Б и им можно пренебречь. В данном случае важно только за какое время автомобиль прошел определенное расстояние и с какой скоростью двигался.
Однако следует учитывать, что пренебрежение размером допускается не в каждой задаче. Например, если рассчитывать движение при парковке автомобиля, то игнорирование величины данного тела, приведет к пагубным последствием. Поэтому, только в тех ситуациях, когда в рамках конкретной задачи, размерами движущегося объекта можно пренебречь, то такое тело принято называть материальной точкой.
Формулы кинематики
Числа, с помощью которых задается положение точки в пространстве, называются координатами. Чтобы определить его на прямой, достаточно одного числа, когда речь идет о поверхности, то двух, о пространстве — трех. Большего количества чисел в трехмерном мире (для описывания положения материальной точки) не требуется.
Существует три основных уравнения для понятия кинематики, как раздела о движении тел:
v = u + at.
S = ut + 1/2at 2 .
v 2 = u 2 + 2as.
v = конечная скорость,
u = Начальная скорость,
a = ускорение,
s = расстояние, пройденное телом,
Формулы кинематики в одномерном пространстве:
X — X o = V o t + 1/2a t2
V 2 = V o 1 + 2a (X — X o)
X — X o = 1\2 (V o + V) t Где,
V — конечная скорость (м / с),
V o — начальная скорость (м / с),
a — ускорение (м / с 2),
t — время (с),
X — конечное положение (м),
Формулы кинематики в двумерном пространстве
Поскольку следующие уравнения используются для описания материальной точки на плоскости, стоит рассматривать ось X и Y.
Учитывая направление Х:
a x = constant
V fx = V i x + a x Δt
X f = X i + V i x Δt +1/2a x Δt 2
Δt = V fx -V ix /a x
V fx 2 = V ix 2 + 2ax Δx
X f = X i + 1/2 (V fx + V ix) Δ t . И учитывая направление y:
a y = constant
V fy = V iy + a y Δt
y f = y i + V iy Δt + 1/2 a x Δt 2
Δt = V fy — V iy /a y
V fy 2 = V iy 2 + 2 ay Δ y
y f = y i +1/2 (V fy + V iy) Δt.
V f — конечная скорость (м / с),
V i — начальная скорость (м / с),
a — ускорение (m / с 2),
t — время (с),
X — конечное положение (м),
X 0 — начальное положение (м).
Перемещение брошенного снаряда — лучший пример для описания движения объекта в двух измерениях. Здесь тело перемещается, как в вертикальном положении У, так и в горизонтальном положении Х, поэтому можно сказать, что предмет имеет две скорости.
Примеры задач по кинематике
Задача 1 : Начальная скорость грузовика равна нулю. Изначально этот объект находится в состоянии покоя. На него начинает действовать равномерное ускорение в течение временного интервала 5,21 секунды. Расстояние, пройденное грузовиком, составляет 110 м. Найти ускорение.
Решение: Пройденное расстояние s = 110 м, начальная скорость v i = 0, время t = 5,21 с, ускорение a =? Используя основные понятие и формулы кинематики, можно заключить, что, s = v i t + 1/2 a t 2 , 110 м = (0) × (5.21) + 1/2 × a (5.21) 2 , a = 8,10 м / с 2 .
Задача 2: Точка движется вдоль оси х (в см), после t секунд путешествия, ее можно представить, используя уравнение x = 14t 2 — t + 10. Необходимо найти среднюю скорость точки, при условии, что t = 3s?
Решение: Положение точки при t = 0, равно x = 10 см. При t = 3s, x = 133 см. Средняя скорость, V av = Δx/Δt = 133-10/3-0 = 41 см / с.
Что такое тело отсчета
О движении можно говорить только если существует что-то, относительно чего рассматривается изменение положения изучаемого объекта. Такой предмет называется телом отсчета и оно условно всегда принимается за неподвижное.
Если в задаче не указано в какой системе отчета движется материальная точка, то телом отсчета считается земля по умолчанию. Однако, это не означает, что за неподвижный в заданный момент времени объект, относительно которого совершается движение, нельзя принять любой другой удобный для расчета. Например, за тело отсчета можно взять движущийся поезд, поворачивающий автомобиль и так далее.
Система отсчета и ее значение в кинематике
Для описания движения необходимы три составляющие:
Система координат.
Тело отсчета.
Прибор для измерения времени.
Тело отсчета, система координат, связанная с ним и прибор для измерения времени образуют систему отсчета. Бессмысленно говорить о движении, если ее не указывать. Правильно подобранная система отсчета, позволяет упростить описание перемещения и, наоборот, усложнить, если она выбрана неудачно.
Именно по этой причине, человечество долго считало, что Солнце движется вокруг Земли и что она находится в центре вселенной. Такое сложное движение светил, связанное с тем, что земные наблюдатели находятся в системе отсчета, которая очень замысловато движется. Земля вращается вокруг свое оси и одновременно вокруг Солнца. На самом деле, если сменить систему отсчета, то все движения небесных тел легко описываются. Это в свое время было сделано Коперником. Он предложил собственное описание мироустройства, в котором Солнце неподвижно. Относительно него описать движение планет гораздо проще, чем если телом отсчета будет являться Земля.
Основные понятия кинематики — путь и траектория
Пусть некоторая точка первое время находилась в положении А, спустя некоторое время она оказалась в положении В. Между ними можно провести одну линию. Но для того, чтобы эта прямая несла больше информации о движении, то есть было понятно откуда и куда двигалось тело, это должен быть не просто отрезок, а направленный, обычно обозначающийся буквой S. Перемещением тела, называется вектор, проведенный из начального положения предмета в конечное.
Если тело изначально находилось в точке А, а затем оказалось в точке В, это не означает, что оно двигалось только по прямой. Из одного положения в другое можно попасть бесконечным количеством способов. Линия, вдоль которой движется тело, является еще одним основным понятием кинематики — траекторией. А ее длина называется путь, который обычно обозначается буквами L или l.
Сессия приближается, и пора нам переходить от теории к практике. На выходных мы сели и подумали о том, что многим студентам было бы неплохо иметь под рукой подборку основных физических формул. Сухие формулы с объяснением: кратко, лаконично, ничего лишнего. Очень полезная штука при решении задач, знаете ли. Да и на экзамене, когда из головы может «выскочить» именно то, что накануне было жесточайше вызубрено, такая подборка сослужит отличную службу.
Больше всего задач обычно задают по трем самым популярным разделам физики. Это механика , термодинамика и молекулярная физика , электричество . Их и возьмем!
Основные формулы по физике динамика, кинематика, статика
Начнем с самого простого. Старое-доброе любимое прямолинейное и равномерное движение.
Формулы кинематики:
Конечно, не будем забывать про движение по кругу, и затем перейдем к динамике и законам Ньютона.
После динамики самое время рассмотреть условия равновесия тел и жидкостей, т.е. статику и гидростатику
Теперь приведем основные формулы по теме «Работа и энергия». Куда же нам без них!
Основные формулы молекулярной физики и термодинамики
Закончим раздел механики формулами по колебаниям и волнам и перейдем к молекулярной физике и термодинамике.
Коэффициент полезного действия, закон Гей-Люссака, уравнение Клапейрона-Менделеева — все эти милые сердцу формулы собраны ниже.
Кстати!
Для всех наших читателей сейчас действует скидка 10% на любой вид работы .
Основные формулы по физике: электричество
Пора переходить к электричеству, хоть его и любят меньше термодинамики. Начинаем с электростатики.
И, под барабанную дробь, заканчиваем формулами для закона Ома, электромагнитной индукции и электромагнитных колебаний.
На этом все. Конечно, можно было бы привести еще целую гору формул, но это ни к чему. Когда формул становится слишком много, можно легко запутаться, а там и вовсе расплавить мозг. Надеемся, наша шпаргалка основных формул по физике поможет решать любимые задачи быстрее и эффективнее. А если хотите уточнить что-то или не нашли нужной формулы: спросите у экспертов студенческого сервиса . Наши авторы держат в голове сотни формул и щелкают задачи, как орешки. Обращайтесь, и вскоре любая задача будет вам «по зубам».
Прежде всего, следует заметить, что речь будет идти о геометрической точке, то есть области пространства, не имеющей размеров. Именно для этого абстрактного образа (модели) и справедливы все представленные ниже определения и формулы. Однако для краткости я в дальнейшем буду часто говорить о движении тела , объекта или частицы . Это я делаю только для того, чтобы Вам легче было читать. Но всегда помните, что речь идет о геометрической точке.
Радиус-вектор точки — это вектор, начало которого совпадает с началом системы координат, а конец — с данной точкой. Радиус-вектор обозначается, как правило, буквой r . К сожалению некоторые авторы обозначают его буквой s . Настоятельно советую не использовать обозначение s для радиус-вектора. Дело в том, что подавляющее большинство авторов (как отечественных, так и зарубежных) используют букву s для обозначения пути, который является скаляром и к радиус-вектору, как правило, отношения не имеет. Если вы будете обозначать радиус-вектор как s , то легко можете запутаться. Еще раз, мы, как и все нормальные люди, будем использовать следующие обозначения: r — радиус-вектор точки, s — путь, пройденный точкой.
Вектор перемещения (часто говорят просто — перемещение ) — это вектор , начало которого совпадает с той точкой траектории, где было тело, когда мы начали изучать данное движение, а конец этого вектора совпадает с той точкой траектории, где мы это изучение закончили. Будем обозначать этот вектор как Δr . Использование символа Δ очевидно: Δr — это разность между радиус-вектором r конечной точки изучаемого отрезка траектории и радиус-вектором r 0 точки начала этого отрезка (рис. 1), то есть Δr = r − r 0 .
Траектория — это линия, вдоль которой движется тело.
Путь — это сумма длин всех участков траектории, последовательно проходимых телом при движения. Обозначается либо ΔS, если речь идет об участке траектории, либо S, если речь идет о всей траектории наблюдаемого движения. Иногда (редко) путь обозначают и другой буквой, например, L (только не обозначайте его как r, мы уже об этом говорили). Запомните! Путь — это положительный скаляр ! Путь в процессе движения может только увеличиваться .
Средняя скорость перемещения v ср
v ср = Δr /Δt.
Мгновенная скорость перемещения v — это вектор, определяемый выражением
v = dr /dt.
Средняя скорость пути v ср — это скаляр, определяемый выражением
V ср = Δs/Δt.
Часто встречаются и другие обозначения, например, .
Мгновенная скорость пути v — это скаляр, определяемый выражением
Модуль мгновенной скорости перемещения и мгновенная скорость пути — это одно и то же, поскольку dr = ds.
Среднее ускорение a
a ср = Δv /Δt.
Мгновенное ускорение (или просто, ускорение ) a — это вектор, определяемый выражением
a =dv /dt.
Касательное (тангенциальное) ускорение a τ (нижний индекс — это греческая строчная буква тау) — это вектор , являющийся векторной проекцией мгновенного ускорения на касательную ось .
Нормальное (центростремительное) ускорение a n — это вектор , являющийся векторной проекцией мгновенного ускорения на ось нормали .
Модуль касательного ускорения
| a τ | = dv/dt,
То есть это — производная модуля мгновенной скорости по времени.
Модуль нормального ускорения
| a n | = v 2 /r,
Где r — величина радиуса кривизны траектории в точке нахождения тела.
Важно! Хочу обратить внимание на следующее. Не путайтесь с обозначениями, касающимися касательного и нормального ускорений! Дело в том, что в литературе по этому поводу традиционно наблюдается полная чехарда.
Запомните!
a τ — это вектор касательного ускорения,
a n — это вектор нормального ускорения.
a τ и a n являются векторными проекциями полного ускорения а на касательную ось и ось нормали соответственно,
A τ — это проекция (скалярная!) касательного ускорения на касательную ось,
A n — это проекция (скалярная!) нормального ускорения на ось нормали,
| a τ |- это модуль вектора касательного ускорения,
| a n | — это модуль вектора нормального ускорения.
Особенно не удивляйтесь, если, читая в литературе о криволинейном (в частности, вращательном) движении, Вы обнаружите, что автор под a τ понимает и вектор, и его проекцию, и его модуль. То же самое относится и к a n . Все, как говорится, «в одном флаконе». И такое, к сожалению, сплошь и рядом. Даже учебники для высшей школы не являются исключением, во многих из них (поверьте — в большинстве!) царит полная неразбериха по этому поводу.
Вот так, не зная азов векторной алгебры или пренебрегая ими, очень легко полностью запутаться при изучении и анализе физических процессов. Поэтому знание векторной алгебры является наиглавнейшим условием успеха в изучении механики. И не только механики. В дальнейшем, при изучении других разделов физики, Вы неоднократно в этом убедитесь.
Мгновенная угловая скорость (или просто, угловая скорость ) ω — это вектор, определяемый выражением
ω = dφ /dt,
Где dφ — бесконечно малое изменение угловой координаты (dφ — вектор!).
Мгновенное угловое ускорение (или просто, угловое ускорение ) ε — это вектор, определяемый выражением
ε = dω /dt.
Связь между v , ω и r :
v = ω × r .
Связь между v, ω и r:
Связь между | a τ |, ε и r:
| a τ | = ε · r.
Теперь перейдем к кинематическим уравнениям конкретных видов движения. Эти уравнения надо выучить наизусть .
Кинематическое уравнение равномерного и прямолинейного движения имеет вид:
r = r 0 + v t,
Где r — радиус-вектор объекта в момент времени t, r 0 — то же в начальный момент времени t 0 (в момент начала наблюдений).
Кинематическое уравнение движения с постоянным ускорением имеет вид:
r = r 0 + v 0 t + a t 2 /2, где v 0 скорость объекта в момент t 0 .
Уравнение для скорости тела при движении с постоянным ускорением имеет вид:
v = v 0 + a t.
Кинематическое уравнение равномерного движения по окружности в полярных координатах имеет вид:
φ = φ 0 + ω z t,
Где φ — угловая координата тела в данный момент времени, φ 0 — угловая координата тела в момент начала наблюдения (в начальный момент времени), ω z — проекция угловой скорости ω на ось Z (обычно эта ось выбирается перпендикулярно плоскости вращения).
Кинематическое уравнение движения по окружности с постоянным ускорением в полярных координатах имеет вид:
φ = φ 0 + ω 0z t + ε z t 2 /2.
Кинематическое уравнение гармонических колебаний вдоль оси X имеет вид:
Х = А Cos (ω t + φ 0),
Где A — амплитуда колебаний, ω — циклическая частота, φ 0 — начальная фаза колебаний.
Проекция скорости точки, колеблющейся вдоль оси X, на эту ось равна:
V x = − ω · A · Sin (ω t + φ 0).
Проекция ускорения точки, колеблющейся вдоль оси X, на эту ось равна:
А x = − ω 2 · A · Cos (ω t + φ 0).
Связь между циклической частотой ω, обычной частотой ƒ и периодом колебаний T:
ω = 2 πƒ = 2 π/T (π = 3,14 — число пи).
Математический маятник имеет период колебаний T, определяемый выражением:
В числителе подкоренного выражения — длина нити маятника, в знаменателе — ускорение свободного падения
Связь между абсолютной v абс, относительной v отн и переносной v пер скоростями:
v абс = v отн + v пер.
Вот, пожалуй, и все определения и формулы, которые могут понадобиться при решении задач на кинематику. Приведенная информация носит только справочный характер и не может заменить электронную книгу, где доступно, подробно и, надеюсь, увлекательно изложена теория этого раздела механики.
Основные единицы измерения величин в системе СИ таковы:
единица измерения длины — метр (1 м),
времени — секунда (1 с),
массы — килограмм (1 кг),
количества вещества — моль (1 моль),
температуры — кельвин (1 К),
силы электрического тока — ампер (1 А),
Справочно: силы света — кандела (1 кд, фактически не используется при решении школьных задач).
При выполнении расчетов в системе СИ углы измеряются в радианах.
Если в задаче по физике не указано, в каких единицах нужно дать ответ, его нужно дать в единицах системы СИ или в производных от них величинах, соответствующих той физической величине, о которой спрашивается в задаче. Например, если в задаче требуется найти скорость, и не сказано в чем ее нужно выразить, то ответ нужно дать в м/с.
Для удобства в задачах по физике часто приходится использовать дольные (уменьшающие) и кратные (увеличивающие) приставки. их можно применять к любой физической величине. Например, мм – миллиметр, кт – килотонна, нс – наносекунда, Мг – мегаграмм, ммоль – миллимоль, мкА – микроампер. Запомните, что в физике не существует двойных приставок. Например, мкг – это микрограмм, а не милликилограмм. Учтите, что при сложении и вычитании величин Вы можете оперировать только величинами одинаковой размерности. Например, килограммы можно складывать только с килограммами, из миллиметров можно вычитать только миллиметры, и так далее. При переводе величин пользуйтесь следующей таблицей.
Путь и перемещение
Кинематикой называют раздел механики, в котором движение тел рассматривается без выяснения причин этого движения.
Механическим движением тела называют изменение его положения в пространстве относительно других тел с течением времени.
Всякое тело имеет определенные размеры. Однако, во многих задачах механики нет необходимости указывать положения отдельных частей тела. Если размеры тела малы по сравнению с расстояниями до других тел, то данное тело можно считать материальной точкой . Так при движении автомобиля на большие расстояния можно пренебречь его длиной, так как длина автомобиля мала по сравнению с расстояниями, которое он проходит.
Интуитивно понятно, что характеристики движения (скорость, траектория и т.д.) зависят от того, откуда мы на него смотрим. Поэтому для описания движения вводится понятие системы отсчета.Система отсчета (СО) – совокупность тела отсчета (оно считается абсолютно твердым), привязанной к нему системой координат, линейки (прибора, измеряющего расстояния), часов и синхронизатора времени.
Перемещаясь с течением времени из одной точки в другую, тело (материальная точка) описывает в данной СО некоторую линию, которую называют траекторией движения тела .
Перемещением тела называют направленный отрезок прямой, соединяющий начальное положение тела с его конечным положением. Перемещение есть векторная величина. Перемещение может в процессе движения увеличиваться, уменьшаться и становиться равным нулю.
Пройденный путь равен длине траектории, пройденной телом за некоторое время. Путь – скалярная величина. Путь не может уменьшаться. Путь только возрастает либо остается постоянным (если тело не движется). При движении тела по криволинейной траектории модуль (длина) вектора перемещения всегда меньше пройденного пути.
При равномерном (с постоянной скоростью) движении путь L может быть найден по формуле:
где: v – скорость тела, t – время в течении которого оно двигалось. При решении задач по кинематике перемещение обычно находится из геометрических соображений. Часто геометрические соображения для нахождения перемещения требуют знания теоремы Пифагора.
Средняя скорость
Скорость – векторная величина, характеризующая быстроту перемещения тела в пространстве. Скорость бывает средней и мгновенной. Мгновенная скорость описывает движение в данный конкретный момент времени в данной конкретной точке пространства, а средняя скорость характеризует все движение в целом, в общем, не описывая подробности движения на каждом конкретном участке.
Средняя скорость пути – это отношение всего пути ко всему времени движения:
где: L полн – весь путь, который прошло тело, t полн – все время движения.
Средняя скорость перемещения – это отношение всего перемещения ко всему времени движения:
Эта величина направлена так же, как и полное перемещение тела (то есть из начальной точки движения в конечную точку). При этом не забывайте, что полное перемещение не всегда равно алгебраической сумме перемещений на определённых этапах движения. Вектор полного перемещения равен векторной сумме перемещений на отдельных этапах движения.
При решении задач по кинематике не совершайте очень распространенную ошибку. Средняя скорость, как правило, не равна среднему арифметическому скоростей тела на каждом этапе движения. Среднее арифметическое получается только в некоторых частных случаях.
И уж тем более средняя скорость не равна одной из скоростей, с которыми двигалось тело в процессе движения, даже если эта скорость имела примерно промежуточное значение относительно других скоростей, с которыми двигалось тело.
Равноускоренное прямолинейное движение
Ускорение – векторная физическая величина, определяющая быстроту изменения скорости тела. Ускорением тела называют отношение изменения скорости к промежутку времени, в течение которого происходило изменение скорости:
где: v 0 – начальная скорость тела, v – конечная скорость тела (то есть спустя промежуток времени t ).
Далее, если иное не указано в условии задачи, мы считаем, что если тело движется с ускорением, то это ускорение остается постоянным. Такое движение тела называется равноускоренным (или равнопеременным). При равноускоренном движении скорость тела изменяется на одинаковую величину за любые равные промежутки времени.
Равноускоренное движение бывает собственно ускоренным, когда тело увеличивает скорость движения, и замедленным, когда скорость уменьшается. Для простоты решения задач удобно для замедленного движения брать ускорение со знаком «–».
Из предыдущей формулы, следует другая более распространённая формула, описывающая изменение скорости со временем при равноускоренном движении:
Перемещение (но не путь) при равноускоренном движении рассчитывается по формулам:
В последней формуле использована одна особенность равноускоренного движения. При равноускоренном движении среднюю скорость можно рассчитывать, как среднее арифметическое начальной и конечной скоростей (этим свойством очень удобно пользоваться при решении некоторых задач):
С расчетом пути все сложнее. Если тело не меняло направления движения, то при равноускоренном прямолинейном движении путь численно равен перемещению. А если меняло – надо отдельно считать путь до остановки (момента разворота) и путь после остановки (момента разворота). А просто подстановка времени в формулы для перемещения в этом случае приведет к типичной ошибке.
Координата при равноускоренном движении изменяется по закону:
Проекция скорости при равноускоренном движении изменяется по такому закону:
Аналогичные формулы получаются для остальных координатных осей.
Свободное падение по вертикали
На все тела, находящиеся в поле тяготения Земли, действует сила тяжести. В отсутствие опоры или подвеса эта сила заставляет тела падать к поверхности Земли. Если пренебречь сопротивлением воздуха, то движение тел только под действием силы тяжести называется свободным падением. Сила тяжести сообщает любым телам, независимо от их формы, массы и размеров, одинаковое ускорение, называемое ускорением свободного падения. Вблизи поверхности Земли ускорение свободного падения составляет:
Это значит, что свободное падение всех тел вблизи поверхности Земли является равноускоренным (но не обязательно прямолинейным) движением. Вначале рассмотрим простейший случай свободного падения, когда тело движется строго по вертикали. Такое движение является равноускоренным прямолинейным движением, поэтому все изученные ранее закономерности и фокусы такого движения подходят и для свободного падения. Только ускорение всегда равно ускорению свободного падения.
Традиционно при свободном падении используют направленную вертикально ось OY. Ничего страшного здесь нет. Просто надо во всех формулах вместо индекса «х » писать «у ». Смысл этого индекса и правило определения знаков сохраняется. Куда направлять ось OY – Ваш выбор, зависящий от удобства решения задачи. Вариантов 2: вверх или вниз.
Приведем несколько формул, которые являются решением некоторых конкретных задач по кинематике на свободное падение по вертикали. Например, скорость, с которой упадет тело падающее с высоты h без начальной скорости:
Время падения тела с высоты h без начальной скорости:
Максимальная высота на которую поднимется тело, брошенное вертикально вверх с начальной скоростью v 0 , время подъема этого тела на максимальную высоту, и полное время полета (до возвращения в исходную точку):
Горизонтальный бросок
При горизонтальном броске с начальной скоростью v 0 движение тела удобно рассматривать как два движения: равномерное вдоль оси ОХ (вдоль оси ОХ нет никаких сил препятствующих или помогающих движению) и равноускоренного движения вдоль оси OY.
Скорость в любой момент времени направлена по касательной к траектории. Ее можно разложить на две составляющие: горизонтальную и вертикальную. Горизонтальная составляющая всегда остается неизменной и равна v x = v 0 . А вертикальная возрастает по законам ускоренного движения v y = gt . При этом полная скорость тела может быть найдена по формулам:
При этом важно понять, что время падения тела на землю никоим образом не зависит от того, с какой горизонтальной скоростью его бросили, а определяется только высотой, с которой было брошено тело. Время падения тела на землю находится по формуле:
Пока тело падает, оно одновременно движется вдоль горизонтальной оси. Следовательно, дальность полета тела или расстояние, которое тело сможет пролететь вдоль оси ОХ, будет равно:
Угол между горизонтом и скоростью тела легко найти из соотношения:
Также иногда в задачах могут спросить о моменте времени, при котором полная скорость тела будет наклонена под определенным углом к вертикали . Тогда этот угол будет находиться из соотношения:
Важно понять, какой именно угол фигурирует в задаче (с вертикалью или с горизонталью). Это и поможет вам выбрать правильную формулу. Если же решать эту задачу координатным методом, то общая формула для закона изменения координаты при равноускоренном движении:
Преобразуется в следующий закон движения по оси OY для тела брошенного горизонтально:
При ее помощи мы можем найти высоту на которой будет находится тело в любой момент времени. При этом в момент падения тела на землю координата тела по оси OY будет равна нулю. Очевидно, что вдоль оси OХ тело движется равномерно, поэтому в рамках координатного метода горизонтальная координата изменятся по закону:
Бросок под углом к горизонту (с земли на землю)
Максимальная высота подъема при броске под углом к горизонту (относительно начального уровня):
Время подъема до максимальной высоты при броске под углом к горизонту:
Дальность полета и полное время полета тела брошенного под углом к горизонту (при условии, что полет заканчивается на той же высоте с которой начался, т.е. тело бросали, например, с земли на землю):
Минимальная скорость тела брошенного под углом к горизонту – в наивысшей точке подъёма, и равна:
Максимальная скорость тела брошенного под углом к горизонту – в моменты броска и падения на землю, и равна начальной. Это утверждение верно только для броска с земли на землю. Если тело продолжает лететь ниже того уровня, с которого его бросали, то оно будет там приобретать все большую и большую скорость.
Сложение скоростей
Движение тел можно описывать в различных системах отсчета. С точки зрения кинематики все системы отсчета равноправны. Однако кинематические характеристики движения, такие как траектория, перемещение, скорость, в разных системах оказываются различными. Величины, зависящие от выбора системы отсчета, в которой производится их измерение, называют относительными. Таким образом, покой и движение тела относительны.
Таким образом, абсолютная скорость тела равна векторной сумме его скорости относительно подвижной системы координат и скорости самой подвижной системы отсчета. Или, другими словами, скорость тела в неподвижной системе отсчета равна векторной сумме скорости тела в подвижной системе отсчета и скорости подвижной системы отсчета относительно неподвижной.
Равномерное движение по окружности
Движение тела по окружности является частным случаем криволинейного движения. Такой вид движения также рассматривается в кинематике. При криволинейном движении вектор скорости тела всегда направлен по касательной к траектории. То же самое происходит и при движении по окружности (см. рисунок). Равномерное движение тела по окружности характеризуется рядом величин.
Период – время, за которое тело, двигаясь по окружности, совершает один полный оборот. Единица измерения – 1 с. Период рассчитывается по формуле:
Частота – количество оборотов, которое совершило тело, двигаясь по окружности, в единицу времени. Единица измерения – 1 об/с или 1 Гц. Частота рассчитывается по формуле:
В обеих формулах: N – количество оборотов за время t . Как видно из вышеприведенных формул, период и частота величины взаимообратные:
При равномерном вращении скорость тела будет определяется следующим образом:
где: l – длина окружности или путь, пройденный телом за время равное периоду T . При движении тела по окружности удобно рассматривать угловое перемещение φ (или угол поворота), измеряемое в радианах. Угловой скоростью ω тела в данной точке называют отношение малого углового перемещения Δφ к малому промежутку времени Δt . Очевидно, что за время равное периоду T тело пройдет угол равный 2π , следовательно при равномерном движении по окружности выполняются формулы:
Угловая скорость измеряется в рад/с. Не забывайте переводить углы из градусов в радианы. Длина дуги l связана с углом поворота соотношением:
Связь между модулем линейной скорости v и угловой скоростью ω :
При движении тела по окружности с постоянной по модулю скоростью изменяется только направление вектора скорости, поэтому движение тела по окружности с постоянной по модулю скоростью является движением с ускорением (но не равноускоренным), так как меняется направление скорости. В этом случае ускорение направлено по радиусу к центру окружности. Его называют нормальным, или центростремительным ускорением , так как вектор ускорения в любой точке окружности направлен к ее центру (см. рисунок).
Модуль центростремительного ускорения связан с линейной v
на этом сайте. Для этого нужно всего ничего, а именно: посвящать подготовке к ЦТ по физике и математике, изучению теории и решению задач по три-четыре часа каждый день. Дело в том, что ЦТ это экзамен, где мало просто знать физику или математику, нужно еще уметь быстро и без сбоев решать большое количество задач по разным темам и различной сложности. Последнему научиться можно только решив тысячи задач.
Выучить все формулы и законы в физике, и формулы и методы в математике . На самом деле, выполнить это тоже очень просто, необходимых формул по физике всего около 200 штук, а по математике даже чуть меньше. В каждом из этих предметов есть около десятка стандартных методов решения задач базового уровня сложности, которые тоже вполне можно выучить, и таким образом, совершенно на автомате и без затруднений решить в нужный момент большую часть ЦТ. После этого Вам останется подумать только над самыми сложными задачами.
Посетить все три этапа репетиционного тестирования по физике и математике. Каждый РТ можно посещать по два раза, чтобы прорешать оба варианта. Опять же на ЦТ, кроме умения быстро и качественно решать задачи, и знания формул и методов необходимо также уметь правильно спланировать время, распределить силы, а главное правильно заполнить бланк ответов, не перепутав ни номера ответов и задач, ни собственную фамилию. Также в ходе РТ важно привыкнуть к стилю постановки вопросов в задачах, который на ЦТ может показаться неподготовленному человеку очень непривычным.
Успешное, старательное и ответственное выполнение этих трех пунктов, а также ответственная проработка итоговых тренировочных тестов , позволит Вам показать на ЦТ отличный результат, максимальный из того, на что Вы способны.
Нашли ошибку?
Если Вы, как Вам кажется, нашли ошибку в учебных материалах, то напишите, пожалуйста, о ней на электронную почту (). В письме укажите предмет (физика или математика), название либо номер темы или теста, номер задачи, или место в тексте (страницу) где по Вашему мнению есть ошибка. Также опишите в чем заключается предположительная ошибка. Ваше письмо не останется незамеченным, ошибка либо будет исправлена, либо Вам разъяснят почему это не ошибка.
Теоретическая механика и кинематика
Механика — это наука о простейших формах движения материи, которые сводятся к простым перемещениям или переходам физических тел с одного положения или состояния в пространстве и времени в другое, в результате взаимодействия между ними.
Теоретическая механика
Механика охватывает целый комплекс дисциплин, изучающих движение и взаимодействие различных материальных тел, например, прикладная механика, гидромеханика, аэромеханическая, небесная механика, биомеханика и др. Изучение наиболее общих свойств движения и взаимодействия всех тел является предметом специальной дисциплины, которую называют теоретическая механика.
Итак, теоретическая механика изучает наиболее общие законы движения и взаимодействия тел, считая своей главной задачей познания количественных и качественных закономерностей, наблюдаемых в природе. С определения теоретической механики следует, что она принадлежит к фундаментальным естественным наукам.
История развития теоретической механики убеждает в том, что она является одной из научных основ техники и технологии, поскольку существует взаимосвязь между проблемами теоретической механики, проблемами техники и технологии.
Теоретическая механика широко применяет такие методы:
абстракции;
обобщение;
математические методы;
методы формальной логики.
Критерием истинности наших знаний является опыт и практика. Таким образом, теоретическая механика имеет дело не с самими материальными объектами, а с их моделями.
Теоретическая механика — это важная наука для подготовки инженерных кадров. Она является основой для изучения таких дисциплин, как:
теория колебаний, гидравлика;
сопротивление материалов;
теория машин, механизмов и тому подобное.
Знание законов теоретической механики, отражающие объективно существующие взаимосвязи, взаимообусловленности механических движений и преобразования энергии, позволяет научно предсказать ход процессов в новых задачах, возникающих при развитии науки, техники и технологии.
Замечание 1
Статикой называется раздел теоретической механики, в котором изучают методы преобразования одних систем сил в другие, эквивалентные им, а также условия равновесия различных систем сил, действующих на твердое тело.
Одним из основных понятий в статике, как и во всей механике, является понятие о силе. Величина, являющаяся мерой механического взаимодействия материальных тел, называется силой. Сила, действующая на тело, является вектором. Она характеризуется точкой приложения, направлением и величиной. В теоретической механике силу принято обозначать $\vec {F} $ cила, $A$- точка приложения силы, прямая $AB$ — линия действия силы.
Рисунок 1. Сила $\vec {F} $. Автор24 — интернет-биржа студенческих работ
В Международной системе единиц (СИ) за единицу силы принимают один ньютон (1Н). Ньютон — это такая сила, которая массе в 1 кг оказывает ускорение в 1 $мс_2$ (1Н = 1кг • м • с-2).
Основные понятия теоретической механики
К основным понятиям теоретической механики, прежде всего, относятся понятия материальной точки и абсолютно твердого тела. Они являются идеальными моделями материальных тел с той или иной степенью абстракции конкретных свойств реальных физических тел.
Определение 1
Материальной точкой называется геометрическая точка, которой приписана определенная масса.
Например, изучая движение планет вокруг Солнца, их рассматривают как материальные точки, в каждой из которых сосредоточена вся масса соответствующей планеты, абстрагируясь при этом от размеров планет.
С понятием материальной точки тесно связано понятие о системе материальных точек.
Определение 2
Абсолютно твердым телом называется тело, которое состоит из системы материальных точек, которые непрерывно заполняют определенную часть пространства таким образом, что расстояние между любыми двумя его точками остается неизменной.
Отметим, что абстракция абсолютно твердого тела позволяет изучать механическое движение тел, не связанных с существующим изменением их формы, в частности с деформацией. Изучение механических движений тел, зависит от их деформируемости, а также движения жидкости и газов, которые приводят к новой абстракции в виде понятие сплошной среды.
Раздел кинематика
Замечание 2
Кинематикой называется раздел теоретической механики, в котором изучается движение системы материальных точек с геометрической точки зрения. Кинематику называют также геометрией движения, поскольку в ней рассматриваются геометрические свойства движения.
Механические движения, что изучаются в кинематике, осуществляются в пространстве и времени. Отметим, что в теоретической механике пространство, в котором происходит движение тел, рассматривается как трехмерное, и все измерения выполняются на основании методов евклидовой геометрии. В механике время считается одинаковым в любых системах отсчета (системах координат) и не зависит от движения этих систем относительно друг друга. Время сказывается буквой $t$ и рассматривается как непрерывная переменная величина, которая применяется в качестве аргумента.
Изучая движение тела, всегда следует знать, в отношении какого другого тела, которое называется телом отсчета, рассматривается это движение. Совокупность тела отсчета, с которым связана система координат, и часов называют системой отсчета. Эта система может быть как подвижной, так и условно неподвижной. Точки тела, постоянно движущиеся, осуществляют в общем случае различные движения. Поэтому, в первую очередь, возникает необходимость изучить движение отдельных точек тела.
Поскольку движение геометрического образа тела будет известным, когда станет известен закон движения всех его точек, определение движения любого геометрического образа предшествует изучению движения одной его точки.
Эта логика лежит в основе разделения кинематики на такие разделы, как кинематика точки и кинематика твердого тела. Для определения положения точки в пространстве выбирают некоторую систему отсчета (систему координат).
Определение 3
Линия, которую описывает точка при своем движении, называется траекторией. Если траектория точки прямая линия, то движение точки называется прямолинейным, если траектория точки кривая, то — криволинейным.
Движение точки относительно выбранной системы отсчета считается заданным, если известно, с помощью которого способа можно определить положение точки в любой момент времени. Основными пространственно-временными (кинематическими) характеристиками движения точки является ее положение, скорость и ускорение.
Исходя из этого, основная задача кинематики точки заключается в нахождении способов задания ее положения и методов определения скорости и ускорения. Движение точки можно определить тремя способами: векторным, координатным и натуральным.
Векторный. Положение точки можно определить с помощью радиус-вектора $\vec {r}$, проведенного с некоторой заданной неподвижной точки $О$ в данную точку $М$. При движении точки радиус-вектор $\vec {r} $меняется по величине и направлению. Каждому моменту времени $t$ соответствует определенное значение $\vec {r}$. 2}$
Основные формулы по физике — МЕХАНИКА
Формулы механики. Механика делится на три раздела: кинематику, динамику и статику. В разделе кинематика рассматриваются такие кинематические характеристики движения, как перемещение, скорость, ускорение. Здесь необходимо использовать аппарат дифференциального и интегрального исчисления.
В основе классической динамики лежат три закона Ньютона. Здесь необходимо обратить внимание на векторный характер действующих на тела сил, входящих в эти законы.
Динамика охватывает такие вопросы, как закон сохранения импульса, закон сохранения полной механической энергии, работа силы.
При изучении кинематики и динамики вращательного движения следует обратить внимание на связь между угловыми и линейными характеристиками. Здесь вводятся понятия момента силы, момента инерции, момента импульса и рассматривается закон сохранения момента импульса.
Смотрите также основные формулы по термодинамике
Таблица основных формул по механике
Физические законы, формулы, переменные
Формулы механики
Скорость мгновенная:
где r — радиус-вектор материальной точки,
t — время;
— производная радиус-вектора материальной точки по времени.
Модуль вектора скорости:
где s — расстояние вдоль траектории движения (путь)
Скорость средняя (модуль):
Ускорение мгновенное:
Модуль вектора ускорения при прямолинейном движении:
Ускорение при криволинейном движении:
1) нормальное
где R — радиус кривизны траектории,
2) тангенциальное
3) полное (вектор)
4) (модуль)
Скорость и путь при движении:
1) равномерном
2) равнопеременном
V0— начальная скорость;
а > 0 при равноускоренном движении;
а < 0 при равнозамедленном движении.
1)
2)
Угловая скорость:
где φ — угловое перемещение.
Угловое ускорение:
Связь между линейными и угловыми величинами:
Импульс материальной точки:
где m — масса материальной точки.
Основное уравнение динамики поступательного движения (II закон Ньютона):
где F — результирующая сила, <>
Формулы сил:
тяжестиP
где g — ускорение свободного падения
трения Fтр
где μ — коэффициент трения,
N — сила нормального давления,
упругости Fупр
где k — коэффициент упругости (жесткости),
Δх — деформация (изменение длины тела).
Закон сохранения импульса для замкнутой системы, состоящей из двух тел:
где — скорости тел до взаимодействия;
— скорости тел после взаимодействия.
Потенциальная энергия тела:
1) поднятого над Землей на высоту h
2) упругодеформированного
1)
2)
Кинетическая энергия поступательного движения:
Работа постоянной силы:
где α — угол между направлением силы и направлением перемещения.
Полная механическая энергия:
Закон сохранения энергии:
силы консервативны
силы неконсервативны
где W1 — энергия системы тел в начальном состоянии;
W2 — энергия системы тел в конечном состоянии.
Момент инерции тел массой m относительно оси, проходящей через центр инерции (центр масс):
1) тонкостенного цилиндра (обруча)
где R — радиус,
2) сплошного цилиндра (диска)
3) шара
4) стержня длиной l, если ось вращения перпендикулярна стержню и проходит через его середину
Момент инерции тела относительно произвольной оси (теорема Штейнера):
где — момент инерции тела относительно оси, проходящей через центр масс, d — расстояние между осями.
Момент силы(модуль):
где l — плечо силы.
Основное уравнение динамики вращательного движения:
где — угловое ускорение,
— результирующий момент сил.
Момент импульса:
1) материальной точки относительно неподвижной точки
где r — плечо импульса,
2) твердого тела относительно неподвижной оси вращения
1)
2)
Закон сохранения момента импульса:
где L1 — момент импульса системы в начальном состоянии,
L2 — момент импульса системы в конечном состоянии.
Кинетическая энергия вращательного движения:
Работа при вращательном движении
где Δφ — изменение угла поворота.
Кинематические уравнения
Целью этого первого раздела «Класса физики» было исследование разнообразных средств, с помощью которых можно описать движение объектов. Разнообразие представлений, которые мы исследовали, включает словесные представления, графические представления, числовые представления и графические представления (графики положения-времени и графики скорости-времени). В Уроке 6 мы исследуем использование уравнений для описания и представления движения объектов.Эти уравнения известны как кинематические уравнения.
Есть множество величин, связанных с движением объектов — смещение (и расстояние), скорость (и скорость), ускорение и время. Знание каждой из этих величин дает описательную информацию о движении объекта. Например, если известно, что автомобиль движется с постоянной скоростью 22,0 м / с, на север в течение 12,0 секунд для смещения на север на 264 метра, то движение автомобиля полностью описано.И если известно, что вторая машина ускоряется из положения покоя с ускорением на восток 3,0 м / с 2 в течение 8,0 секунд, обеспечивая конечную скорость 24 м / с, восток и смещение на восток 96 метров. , то полностью описывается движение этой машины. Эти два утверждения дают полное описание движения объекта. Однако не всегда такая полнота известна. Часто бывает так, что известны лишь некоторые параметры движения объекта, а остальные неизвестны.Например, приближаясь к светофору, вы можете узнать, что ваша машина развивает скорость 22 м / с, восток и способна выдерживать заносное ускорение 8,0 м / с 2 , запад. Однако вы не знаете, какое смещение испытает ваша машина, если бы вы резко нажали на тормоз и занесло до полной остановки; и вы не знаете, сколько времени потребуется, чтобы остановиться. В таком случае неизвестные параметры могут быть определены с использованием физических принципов и математических уравнений (кинематических уравнений).
БОЛЬШОЙ 4
Кинематические уравнения — это набор из четырех уравнений, которые можно использовать для прогнозирования неизвестной информации о движении объекта, если известна другая информация. Уравнения можно использовать для любого движения, которое можно описать как движение с постоянной скоростью (ускорение 0 м / с / с) или движение с постоянным ускорением. Их нельзя использовать в течение какого-либо периода времени, в течение которого изменяется ускорение.Каждое из кинематических уравнений включает четыре переменные. Если известны значения трех из четырех переменных, то можно рассчитать значение четвертой переменной. Таким образом, кинематические уравнения предоставляют полезные средства прогнозирования информации о движении объекта, если известна другая информация. Например, если известно значение ускорения, а также начальное и конечное значения скорости буксирующего автомобиля, то смещение автомобиля и время можно предсказать с помощью кинематических уравнений.Урок 6 этого модуля будет посвящен использованию кинематических уравнений для прогнозирования числовых значений неизвестных величин для движения объекта.
Четыре кинематических уравнения, описывающие движение объекта:
В приведенных выше уравнениях используются различные символы. Каждый символ имеет свое особое значение. Символ d обозначает смещение объекта. Символ t обозначает время, в течение которого объект двигался.Символ a обозначает ускорение объекта. А символ v обозначает скорость объекта; индекс i после v (как в v i ) указывает, что значение скорости является начальным значением скорости, а индекс f (как в v f ) указывает, что значение скорости является конечным значением скорости.
Каждое из этих четырех уравнений надлежащим образом описывает математическую связь между параметрами движения объекта. Таким образом, они могут использоваться для прогнозирования неизвестной информации о движении объекта, если известна другая информация.В следующей части Урока 6 мы исследуем процесс этого.
Кинематические уравнения и решение проблем
Четыре кинематических уравнения, которые описывают математическую связь между параметрами, описывающими движение объекта, были введены в предыдущей части Урока 6. Четыре кинематических уравнения:
В приведенных выше уравнениях символ d обозначает смещение объекта.Символ t обозначает время, в течение которого объект двигался. Символ a обозначает ускорение объекта. А символ v обозначает мгновенную скорость объекта; индекс i после v (как в v i ) указывает, что значение скорости является начальным значением скорости, а индекс f (как в v f ) указывает, что значение скорости является конечным значением скорости.
Стратегия решения проблем
В этой части Урока 6 мы исследуем процесс использования уравнений для определения неизвестной информации о движении объекта.Процесс включает использование стратегии решения проблем, которая будет использоваться на протяжении всего курса. Стратегия предполагает следующие шаги:
Постройте информативную диаграмму физической ситуации.
Определите и перечислите данную информацию в переменной форме.
Определите и перечислите неизвестную информацию в переменной форме.
Определите и перечислите уравнение, которое будет использоваться для определения неизвестной информации из известной информации.
Подставьте известные значения в уравнение и используйте соответствующие алгебраические шаги, чтобы найти неизвестную информацию.
Проверьте свой ответ, чтобы убедиться, что он разумный и математически правильный.
Использование этой стратегии решения проблем при решении следующей проблемы смоделировано в примерах A и B ниже.
Пример задачи A
Има Харрин приближается к светофору, движущемуся со скоростью +30.0 м / с. Загорается желтый свет, и Има тормозит и останавливается. Если ускорение Има составляет -8,00 м / с 2 , то определите смещение автомобиля во время заноса. (Обратите внимание, что направление векторов скорости и ускорения обозначено знаками «+» и «-».)
Решение этой проблемы начинается с построения информативной диаграммы физической ситуации. Это показано ниже. Второй шаг включает идентификацию и перечисление известной информации в переменной форме.Обратите внимание, что значение v f может быть принято равным 0 м / с, поскольку машина Имы останавливается. Начальная скорость (v i ) кабины +30,0 м / с, так как это скорость в начале движения (заносное движение). А ускорение (а) автомобиля определяется как — 8,00 м / с 2 . (Всегда обращайте особое внимание на знаки + и — для данных количеств.) Следующий шаг стратегии включает перечисление неизвестной (или желаемой) информации в переменной форме.В этом случае проблема запрашивает информацию о перемещении автомобиля. Итак, d — неизвестная величина. Результаты первых трех шагов показаны в таблице ниже.
Схема:
Дано:
Находка:
v i = +30,0 м / с v f = 0 м / с
a = — 8,00 м / с 2
d = ??
Следующий шаг стратегии включает определение кинематического уравнения, которое позволит вам определить неизвестную величину.На выбор предлагается четыре кинематических уравнения. В общем, вы всегда будете выбирать уравнение, которое содержит три известные и одну неизвестную переменные. В этом конкретном случае три известные переменные и одна неизвестная переменная: v f , v i , a и d. Таким образом, вы будете искать уравнение, в котором перечислены эти четыре переменные. Анализ четырех приведенных выше уравнений показывает, что уравнение в правом верхнем углу содержит все четыре переменные.
v f 2 = v i 2 + 2 • a • d
После того, как уравнение идентифицировано и записано, следующий шаг стратегии включает в себя замену известных значений в уравнение и использование соответствующих алгебраических шагов для поиска неизвестной информации.Этот шаг показан ниже.
(0 м / с) 2 = (30,0 м / с) 2 + 2 • (-8,00 м / с 2 ) • d
0 м 2 / с 2 = 900 м 2 / с 2 + (-16,0 м / с 2 ) • d
(16,0 м / с 2 ) • d = 900 м 2 / с 2 — 0 м 2 / с 2
(16,0 м / с 2 ) * d = 900 м 2 / с 2
d = (900 м 2 / с 2 ) / (16.0 м / с 2 )
d = (900 м 2 / с 2 ) / (16,0 м / с 2 )
d = 56,3 м
Решение, приведенное выше, показывает, что автомобиль заносит расстояние 56,3 метра. (Обратите внимание, что это значение округлено до третьей цифры.)
Последний шаг стратегии решения проблем включает проверку ответа, чтобы убедиться, что он является одновременно разумным и точным. Стоимость кажется достаточно разумной. Машине требуется значительное расстояние, чтобы занести из 30.0 м / с (примерно 65 миль / ч) до остановки. Расчетное расстояние составляет примерно половину футбольного поля, что делает его очень разумным расстоянием для заноса. Проверка точности включает подстановку вычисленного значения обратно в уравнение для смещения и обеспечение того, чтобы левая часть уравнения была равна правой части уравнения. В самом деле!
Пример задачи B
Бен Рушин ждет на светофоре. Когда он наконец стал зеленым, Бен ускорился из состояния покоя со скоростью 6,00 м / с 2 за время 4,10 секунды. Определите перемещение машины Бена за этот период времени.
И снова решение этой проблемы начинается с построения информативной диаграммы физической ситуации. Это показано ниже. Второй шаг стратегии включает идентификацию и перечисление известной информации в переменной форме. Обратите внимание, что значение v i можно вывести как 0 м / с, поскольку машина Бена изначально находится в состоянии покоя.Ускорение (а) автомобиля составляет 6,00 м / с 2 . Время (t) равно 4,10 с. Следующий шаг стратегии включает перечисление неизвестной (или желаемой) информации в переменной форме. В этом случае проблема запрашивает информацию о перемещении автомобиля. Итак, d — неизвестная информация. Результаты первых трех шагов показаны в таблице ниже.
Схема:
Дано:
Находка:
v i = 0 м / с т = 4.10 с
a = 6,00 м / с 2
d = ??
Следующий шаг стратегии включает определение кинематического уравнения, которое позволит вам определить неизвестную величину. На выбор предлагается четыре кинематических уравнения. Опять же, вы всегда будете искать уравнение, которое содержит три известные переменные и одну неизвестную переменную. В этом конкретном случае три известные переменные и одна неизвестная переменная — это t, v i , a и d.Анализ четырех приведенных выше уравнений показывает, что уравнение в левом верхнем углу содержит все четыре переменные.
d = v i • t + ½ • a • t 2
После того, как уравнение идентифицировано и записано, следующий шаг стратегии включает в себя замену известных значений в уравнение и использование соответствующих алгебраических шагов для поиска неизвестной информации. Этот шаг показан ниже.
d = (0 м / с) • (4.1 с) + ½ • (6,00 м / с 2 ) • (4,10 с) 2
d = (0 м) + ½ • (6,00 м / с 2 ) • (16,81 с 2 )
d = 0 м + 50,43 м
d = 50,4 м
Решение, приведенное выше, показывает, что автомобиль преодолеет расстояние 50,4 метра. (Обратите внимание, что это значение округлено до третьей цифры.)
Последний шаг стратегии решения проблем включает проверку ответа, чтобы убедиться, что он является одновременно разумным и точным.Стоимость кажется достаточно разумной. Автомобиль с ускорением 6,00 м / с / с достигнет скорости примерно 24 м / с (примерно 50 миль / ч) за 4,10 с. Расстояние, на которое такая машина будет перемещена в течение этого периода времени, будет примерно половиной футбольного поля, что делает это расстояние очень разумным. Проверка точности включает подстановку вычисленного значения обратно в уравнение для смещения и обеспечение того, чтобы левая часть уравнения была равна правой части уравнения.В самом деле!
Два приведенных выше примера задач иллюстрируют, как кинематические уравнения могут быть объединены с простой стратегией решения проблем для прогнозирования неизвестных параметров движения для движущегося объекта. Если известны три параметра движения, можно определить любое из оставшихся значений. В следующей части Урока 6 мы увидим, как эту стратегию можно применить к ситуациям свободного падения. Или, если интересно, вы можете попробовать несколько практических задач и сравнить свой ответ с данными решениями.
Решение проблем базовой кинематики | Безграничная физика
Приложения
Есть четыре кинематических уравнения, которые описывают движение объектов без учета его причин.
Цели обучения
Выберите, какое уравнение кинематики использовать в задачах, в которых начальное начальное положение равно нулю
Основные выводы
Ключевые моменты
Четыре кинематических уравнения включают пять кинематических переменных: [latex] \ text {d} [/ latex], [latex] \ text {v} [/ latex], [latex] \ text {v} _0 [/ latex] , [латекс] \ text {a} [/ latex] и [латекс] \ text {t} [/ latex].
Каждое уравнение содержит только четыре из пяти переменных, а другая отсутствует.
Важно выбрать уравнение, которое содержит три известные переменные и одну неизвестную переменную для каждой конкретной ситуации.
Ключевые термины
кинематика : Раздел физики, связанный с движущимися объектами.
Кинематика — это раздел классической механики, который описывает движение точек, тел (объектов) и систем тел (групп объектов) без учета причин движения.2 + 2 \ text {ad} [/ latex]
Обратите внимание, что четыре кинематических уравнения включают пять кинематических переменных: [latex] \ text {d} [/ latex] , [latex] \ text {v} [/ latex] , [latex] \ text {v } _0 [/ latex] , [латекс] \ text {a} [/ latex] и [латекс] \ text {t} [/ latex]. Каждое из этих уравнений содержит только четыре из пяти переменных, а другая отсутствует. Это говорит нам, что нам нужны значения трех переменных, чтобы получить значение четвертой, и нам нужно выбрать уравнение, которое содержит три известные переменные и одну неизвестную переменную для каждой конкретной ситуации.
Вот основные этапы решения проблем с использованием этих уравнений:
Шаг первый — Определите, что именно необходимо определить в проблеме (определите неизвестные).
Шаг второй. Найдите уравнение или систему уравнений, которые помогут вам решить проблему.
Шаг третий — Подставьте известные значения вместе с их единицами измерения в соответствующее уравнение и получите численные решения вместе с единицами измерения.
Шаг четвертый. Проверьте ответ, чтобы узнать, разумен ли он: имеет ли он смысл?
Навыки решения проблем, безусловно, необходимы для успешного прохождения количественного курса физики.Что еще более важно, способность применять общие физические принципы, обычно представленные уравнениями, к конкретным ситуациям — очень мощная форма знания. Это намного эффективнее, чем запоминание списка фактов. Аналитические навыки и способности решать проблемы могут быть применены к новым ситуациям, тогда как список фактов не может быть достаточно длинным, чтобы содержать все возможные обстоятельства. Такие аналитические навыки полезны как для решения задач на уроках физики, так и для применения физики в повседневной и профессиональной жизни.
Диаграммы движения
Диаграмма движения — это графическое описание движения объекта, которое представляет положение объекта через равные промежутки времени.
Цели обучения
Построить диаграмму движения
Основные выводы
Ключевые моменты
Диаграммы движения представляют движение объекта путем отображения его местоположения в разное время с равным интервалом на одной диаграмме.
Диаграммы движения показывают начальное положение и скорость объекта, а также несколько точек в центре диаграммы.Эти пятна показывают состояние движения объекта.
Диаграммы движения содержат информацию о положении объекта в определенные моменты времени и поэтому являются более информативными, чем диаграмма путей.
Ключевые термины
стробоскопический : Относится к инструменту, который заставляет циклически движущийся объект казаться медленно движущимся или неподвижным.
диаграмма : график или диаграмма.
движение : изменение положения относительно времени.
Диаграмма движения — это графическое описание движения объекта. Он отображает местоположение объекта в разное время с равным интервалом на одной диаграмме; показывает начальное положение и скорость объекта; и представляет несколько точек в центре диаграммы. Эти пятна показывают, ускорился или замедлился объект. Для простоты объект представлен простой формой, например закрашенным кружком, который содержит информацию о положении объекта в определенные моменты времени. По этой причине диаграмма движения дает больше информации, чем диаграмма пути. Он также может отображать силы, действующие на объект в каждый момент времени.
— диаграмма движения по простой траектории. Представьте себе объект в виде хоккейной шайбы, скользящей по льду. Обратите внимание, что шайба преодолевает одинаковое расстояние за единицу пути по траектории. Можно сделать вывод, что шайба движется с постоянной скоростью и, следовательно, во время движения нет ускорения или замедления.
Шайба, скользящая по льду : Диаграмма движения шайбы, скользящей по льду.Шайба движется с постоянной скоростью.
Одно из основных применений диаграмм движения — это представление фильма через серию кадров, снятых камерой; это иногда называют стробоскопической техникой (как показано на рисунке). Просмотр объекта на диаграмме движения позволяет определить, ускоряется или замедляется объект или находится в постоянном покое. Когда кадры сделаны, мы можем предположить, что объект находится в постоянном покое, если он занимает одно и то же положение с течением времени. Мы можем предположить, что объект ускоряется, если есть видимое увеличение пространства между объектами с течением времени, и что он замедляется, если есть видимое уменьшение пространства между объектами с течением времени.Объекты на кадре очень близко подходят друг к другу.
прыгающий мяч : прыгающий мяч, снятый с помощью стробоскопической вспышки со скоростью 25 изображений в секунду.
Формула кинематических уравнений
Кинематика — это исследование движущихся объектов и их взаимосвязей. Есть четыре (4) кинематических уравнения, которые относятся к смещению D, скорости v, времени t и ускорению a.
a) D = v i t + 1/2 при 2 b) (v i + v f ) / 2 = D / t
c) a = (v f — v i ) / t d) v f 2 = v i 2 + 2aD
D = смещение
a = ускорение
т = время
v f = конечная скорость
v i = начальная скорость
Формула кинематических уравнений.
1) Боб едет на велосипеде в магазин со скоростью 4 м / с, когда перед ним выбегает кошка. Он быстро тормозит до полной остановки, с ускорением — 2м / с 2 . Какое у него перемещение?
Ответ: Поскольку Боб остановлен, конечная скорость v f = 0. Его начальная скорость v i = 4 м / с. Ускорение, a = -2 м / с 2 . Время не указано, поэтому используйте уравнение (d) для смещения D, потому что оно не зависит от времени.
v f 2 = v i 2 + 2aD
(0) 2 = (4 м / с) 2 +2 (- 2 м / с 2 ) D
0 = 16 м 2 / с 2 + (- 4 м / с 2 ) D
-16 м 2 / с 2 = (- 4 м / с 2 ) D
16 м 2 / с 2 = 4 м / с 2 ) D
(16 м 2 / с 2 ) / (4 м / с 2 ) = D
Водоизмещение полное 4 м.
2) Вы путешествуете с постоянной скоростью 11 м / с в течение 5 минут. Как далеко ты проехал?
Ответ: При постоянной скорости v i = v f = 11 м / с. Время t = 5 мин или t = (60 сек / мин x 5 мин) = 300 сек. Теперь используйте уравнение (b), чтобы найти смещение D.
(v i + v f ) / 2 = D / t
D = [(v i + v f ) / 2] t
D = [(11 м / с + 11 м / с) / 2] x 300 с
D = (22 м / с) / 2 x 300 с
D = 11 м / с x 300 с
D = 3300 м Водоизмещение полное 3,300 м.
3) Каково ускорение автомобиля, который разгоняется с 11 до 40 м / с за 10 секунд?
Ответ: V i = 11 м / с. V f = 40 м / с. Время, t = 10 с. Используйте кинематическое уравнение c), чтобы найти ускорение.
a = (v f — v i ) / t
a = (40 м / с — 11 м / с) / 10 с
a = (29 м / с) / 10 с = 2,9 м / с 2
4) Если автомобиль разгоняется на 3. 0 м / с 2 от полной остановки, сколько времени потребуется, чтобы проехать 3000 м?
Ответ: Ускорение a = 2,9 м / с 2 и перемещение D = 3000 м. Автомобиль был неподвижен, поэтому v i = 0. Используйте уравнение a), чтобы найти время.
D = v i t + 1/2 при 2
3000 м = 0т + 1/2 (3,0 м / с 2 ) т 2
3000 м = 1/2 (3,0 м / с 2 ) / т 2
3000 м / 1.5 м / с 2 = t 2
2000 с 2 = t 2
t = 44,72 сек
Каковы кинематические формулы?
Kinematic Equations: Целью этого первого раздела The Physics Classroom было исследование разнообразных средств, с помощью которых может быть описано движение объектов. Разнообразие представлений, которые мы исследовали, включает словесные представления, графические представления, числовые представления и графические представления (графики положения-времени и графики скорости-времени).
В Уроке 6 мы исследуем использование уравнений для описания и представления движения объектов. Эти уравнения известны как кинематические уравнения. Существует множество величин, связанных с движением объектов: смещение (и расстояние), скорость (и скорость), ускорение и время.
Что такое кинематические формулы?
Знание каждой из этих величин дает описательную информацию о движении объекта. Например, если известно, что автомобиль движется с постоянной скоростью 22.0 м / с, север в течение 12,0 секунд для смещения на север на 264 метра, затем движение автомобиля полностью описывается. И если известно, что вторая машина ускоряется из положения покоя с ускорением на восток 3,0 м / с 2 в течение 8,0 секунд, обеспечивая конечную скорость 24 м / с, восток и смещение на восток 96 метров. , то полностью описывается движение этой машины.
Эти два утверждения дают полное описание движения объекта. Однако не всегда такая полнота известна.Часто бывает так, что известны лишь некоторые параметры движения объекта, а остальные неизвестны. Например, приближаясь к светофору, вы можете узнать, что ваша машина имеет скорость 22 м / с, восток, и способна к заносу 8,0 м / с 2 , запад.
Однако вы не знаете, какое смещение испытает ваша машина, если бы вы резко нажали на тормоз и занесло до полной остановки; и вы не знаете, сколько времени потребуется, чтобы остановиться. В таком случае неизвестные параметры могут быть определены с использованием принципов физики и математических уравнений (кинематических уравнений)
4 кинематических уравнения
Кинематика — это изучение движущихся объектов и их взаимосвязей.Есть четыре (4) кинематических уравнения, которые относятся к смещению D, скорости v, времени t и ускорению a.
a) D = v i t + 1/2 при 2 b) (v i + v f ) / 2 = D / t
c) a = (v f — v i ) / t d) v f 2 = v i 2 + 2aD
D = смещение
a = ускорение
т = время
v f = конечная скорость
v i = начальная скорость
Какие 3 кинематических уравнения?
Если мы знаем три из этих пяти кинематических переменных — Δ x, t, v 0, v, a \ Delta x, t, v_0, v, a Δx, t, v0, v, adelta, x, запятая , t, запятая, v, начальный индекс, 0, конечный индекс, запятая, v, запятая, a — для объекта с постоянным ускорением мы можем использовать кинематическую формулу , см. ниже, чтобы найти одну из неизвестных переменных .
Сколько существует кинематических уравнений?
Четыре кинематических уравнения , которые описывают движение объекта: В приведенных выше уравнениях используются различные символы. Каждый символ имеет свое особое значение. Символ d обозначает смещение объекта.
Для чего используются кинематические уравнения?
Кинематические уравнения могут быть использованы для вычисления различных аспектов движения, таких как скорость, ускорение, смещение и время.
Список кинематических уравнений
Кинематические формулы — это набор формул, которые относятся к пяти кинематическим переменным, перечисленным ниже.
Большой 1.
четырехъядерный v = v0 + at1
v = v0 + at1,
точка, пробел, v, равно, v, начальный индекс, 0, конечный индекс, плюс, a, t
Поскольку кинематические формулы являются точными только в том случае, если ускорение является постоянным в течение рассматриваемого интервала времени, мы должны быть осторожны, чтобы не использовать их при изменении ускорения. Кроме того, кинематические формулы предполагают, что все переменные относятся к одному и тому же направлению: по горизонтали xxx, по вертикали yyy и т. Д.2} g = 9,81s2m g, равно, 9, точка, 81, начальная дробь, m, деленная на, s, начальный верхний индекс, 2, конечный верхний индекс, конечная дробь.
Это и странно, и удачно, если вдуматься. Это странно, так как это означает, что большой валун будет ускоряться вниз с таким же ускорением, как и небольшой камешек, и, если его уронить с той же высоты, он одновременно ударится о землю.
Это удачно, поскольку нам не нужно знать массу снаряда при решении кинематических формул, поскольку свободно летящий объект будет иметь такую же величину ускорения, g = 9.2} ay = −9,81s2m a, начальный индекс, y, конечный индекс, равно, минус, 9, точка, 81, начальная дробь, m, деленное на, s, начальный верхний индекс, 2, конечный верхний индекс, конечная дробь для снаряд, когда мы подставляем кинематические формулы.
Физика кинематических уравнений
Кинематические уравнения — это набор из четырех уравнений, которые можно использовать для прогнозирования неизвестной информации о движении объекта, если известна другая информация. Уравнения можно использовать для любого движения, которое можно описать как движение с постоянной скоростью (ускорение 0 м / с / с) или движение с постоянным ускорением.Их нельзя использовать в течение какого-либо периода времени, в течение которого изменяется ускорение. Каждое из кинематических уравнений включает четыре переменные. Если известны значения трех из четырех переменных, то можно рассчитать значение четвертой переменной. Таким образом, кинематические уравнения предоставляют полезные средства прогнозирования информации о движении объекта, если известна другая информация.
Например, если значение ускорения, а также начальное и конечное значения скорости буксирующего автомобиля известны, то смещение автомобиля и время можно предсказать с помощью кинематических уравнений.Урок 6 этого модуля будет посвящен использованию кинематических уравнений для прогнозирования числовых значений неизвестных величин для движения объекта.
Четыре кинематических уравнения, описывающие движение объекта:
В приведенных выше уравнениях используются различные символы. Каждый символ имеет свое особое значение. Символ d обозначает смещение объекта. Символ t обозначает время, в течение которого объект двигался. Символ обозначает ускорение объекта.А символ v обозначает скорость объекта; индекс i после v (как в v i ) указывает, что значение скорости является начальным значением скорости, а индекс f (как в v f ) указывает, что значение скорости является конечным значением скорости.
Каждое из этих четырех уравнений надлежащим образом описывает математическую связь между параметрами движения объекта. Таким образом, их можно использовать для прогнозирования неизвестной информации о движении объекта, если известна другая информация.В следующей части Урока 6 мы исследуем, как это сделать.
Читайте также: Формула линейной интерполяции
Основные кинематические уравнения
Кинематика — это исследование движения объектов без учета сил, вызывающих это движение. Эти знакомые уравнения позволяют студентам анализировать и предсказывать движение объектов, и студенты будут продолжать использовать эти уравнения на протяжении всего изучения физики. Твердое понимание этих уравнений и того, как их использовать для решения проблем, необходимо для успеха в физике.Эта статья представляет собой чисто математическое упражнение, предназначенное для быстрого обзора того, как уравнения кинематики выводятся с помощью алгебры.
Это упражнение ссылается на диаграмму на рис. 1, на которой ось x представляет время, а ось y представляет скорость. Диагональная линия представляет движение объекта, скорость которого изменяется с постоянной скоростью. Заштрихованная область (A 1 + A 2 ) представляет смещение объекта в течение интервала времени между t 1 и t 2 , в течение которого скорость объекта увеличилась с v 1 до v 2. .
В этом документе будут использоваться следующие переменные: v = величина скорости объекта (метры в секунду, м / с) v 1 = величина начальной скорости (метров на секунда, м / с) (в некоторых текстах это vi или v 0 ) v 2 = величина конечной скорости (метры в секунду, м / с) (в некоторых текстах это v f ) a = величина ускорения (в метрах в секунду в квадрате, м / с 2 ) с = вектор смещения, величина смещения — это расстояние, с = │ s │ = d (векторы выделены жирным шрифтом; тот же символ, не выделенный жирным шрифтом, представляет величину вектора) Δ указывает на изменение, например Δv = (v 2 –v 1 ) t = время t 1 = начальное время t 2 = Последний раз
Equations of Motion — The Physics Hypertextbook
Обсуждение
постоянное ускорение
Для точности этот раздел должен называться «Одномерные уравнения движения при постоянном ускорении». Учитывая, что такое название было бы стилистическим кошмаром, позвольте мне начать этот раздел со следующей оговорки. Эти уравнения движения действительны только тогда, когда ускорение постоянное и движение ограничено прямой линией.
Учитывая, что мы живем в трехмерной вселенной, в которой единственная константа — это изменение, у вас может возникнуть соблазн сразу отказаться от этого раздела. Было бы правильно сказать, что ни один объект никогда не двигался по прямой с постоянным ускорением в любом месте Вселенной в любое время — ни сегодня, ни вчера, ни завтра, ни пять миллиардов лет назад, ни тридцать миллиардов лет в будущем. , никогда.Об этом я могу сказать с абсолютной метафизической уверенностью.
Так что же тогда хорошего в этом разделе? Что ж, во многих случаях полезно предположить, что объект двигался или будет двигаться по прямому пути с почти постоянным ускорением; то есть любое отклонение от идеального движения можно по существу игнорировать. Движение по криволинейной траектории можно считать фактически одномерным, если для задействованных объектов существует только одна степень свободы .Дорога может изгибаться и поворачиваться и исследовать всевозможные направления, но автомобили, движущиеся по ней, имеют только одну степень свободы — свободу двигаться в одном или противоположном направлении. (Вы не можете двигаться по дороге по диагонали и надеетесь остаться на ней надолго.) В этом отношении это мало чем отличается от движения, ограниченного прямой линией. Аппроксимация реальных ситуаций моделями, основанными на идеальных ситуациях, не считается обманом. Так поступают в физике. Это настолько полезный метод, что мы будем использовать его снова и снова.
Наша цель в этом разделе — вывести новые уравнения, которые можно использовать для описания движения объекта в терминах его трех кинематических переменных: скорости ( v ), положения ( с ) и времени ( т ). Есть три способа объединить их в пары: скорость-время, положение-время и скорость-положение. В этом порядке их также часто называют первым, вторым и третьим уравнениями движения, но нет веских причин для изучения этих имен.
Поскольку мы имеем дело с движением по прямой линии, направление будет обозначено знаком — положительные величины указывают в одну сторону, а отрицательные величины указывают в противоположную сторону.Определение того, какое направление является положительным, а какое отрицательным, совершенно произвольно. Законы физики изотропны ; то есть они не зависят от ориентации системы координат. Однако некоторые проблемы легче понять и решить, если одно направление предпочтительнее другого. Пока вы последовательны в решении проблемы, это не имеет значения.
скорость-время
Связь между скоростью и временем проста при равномерно ускоренном прямолинейном движении.Чем дольше ускорение, тем больше изменение скорости. Изменение скорости прямо пропорционально времени, когда ускорение постоянно. Если скорость увеличивается на определенную величину за определенное время, она должна увеличиваться вдвое на эту величину в два раза быстрее. Если объект уже стартовал с определенной скоростью, то его новая скорость будет равна старой скорости плюс это изменение. Вы должны быть в состоянии увидеть уравнение уже мысленным взором.
Это самое простое из трех уравнений, которое можно вывести с помощью алгебры.Начнем с определения ускорения.
Расширить ∆ v до v — v 0 и сжать ∆ t до t .
Затем найдите v как функцию от t .
v = v 0 + at [1]
Это первое уравнение движения . Он записывается как полином — постоянный член ( v 0 ), за которым следует член первого порядка ( на ).Поскольку наивысший порядок равен 1, правильнее называть его линейной функцией 1.
Символ v 0 [vee naught] называется начальной скоростью или скоростью t = 0. Его часто называют «первой скоростью», но это довольно наивный способ Опишите это. Лучшее определение было бы сказать, что начальная скорость — это скорость, которую имеет движущийся объект, когда он впервые становится важным в проблеме. Скажем, метеор был замечен глубоко в космосе, и проблема заключалась в том, чтобы определить его траекторию, тогда начальная скорость, вероятно, будет той скоростью, которую он имел при первом наблюдении.Но если проблема заключалась в том, что тот же самый метеор сгорает при входе в атмосферу, то начальная скорость, вероятно, равна скорости, которую он имел при входе в атмосферу Земли. Ответ на вопрос «Какая начальная скорость?» «Это зависит от обстоятельств». Это оказывается ответом на множество вопросов.
Символ v — это скорость через некоторое время t после начальной скорости. Ее часто называют конечной скоростью , но это не делает ее «последней скоростью» объекта. Возьмем случай с метеором.Какая скорость обозначена символом v ? Если вы внимательно слушали, значит, вы должны были ожидать ответа. По-разному. Это может быть скорость метеора, когда он проходит мимо Луны, входит в атмосферу Земли или ударяется о поверхность Земли. Это также может быть скорость метеорита, находящегося на дне кратера. (В этом случае v = 0 м / с.) Является ли какое-либо из этих значений конечной скоростью? Кто знает. Кто-то мог извлечь метеорит из дыры в земле и уехать вместе с ним.Это актуально? Наверное, нет, но это зависит от обстоятельств. Для такого рода вещей нет правил. Вы должны проанализировать текст задачи на предмет физических величин, а затем присвоить значение математическим символам.
Последняя часть этого уравнения на — это изменение скорости от начального значения. Вспомните, что a — это скорость изменения скорости, а t — это время после некоторого начального события . Ставка раз время меняется. Учитывая, что объект ускоряется со скоростью 10 м / с 2 , через 5 с он будет двигаться на 50 м / с быстрее. Если бы он стартовал со скоростью 15 м / с, то его скорость через 5 с была бы…
15 м / с + 50 м / с = 65 м / с
время позиции
Смещение движущегося объекта прямо пропорционально скорости и времени. Двигайся быстрее. Иди дальше. Двигайтесь дольше (как и дольше). Иди дальше. Ускорение усугубляет эту простую ситуацию, поскольку скорость теперь также прямо пропорциональна времени. Попробуйте сказать это словами, и это прозвучит нелепо. «Смещение прямо пропорционально времени и прямо пропорционально скорости, которая прямо пропорциональна времени.»Время удваивается, поэтому смещение пропорционально квадрату времени. Автомобиль, разгоняющийся в течение двух секунд, преодолеет в четыре раза больше расстояния, чем автомобиль, ускоряющийся всего за одну секунду (2 2 = 4). Автомобиль, ускоряющийся в течение трех секунды покрыли бы расстояние в девять раз большее (3 2 = 9).
Если бы это было так просто. Этот пример работает, только когда начальная скорость равна нулю. Смещение пропорционально квадрату времени, когда ускорение постоянное, а начальная скорость равна нулю.Истинное общее утверждение должно учитывать любую начальную скорость и то, как она менялась. Это приводит к ужасно запутанному утверждению соразмерности. Смещение прямо пропорционально времени и пропорционально квадрату времени, когда ускорение постоянно. Функция, которая является одновременно линейной и квадратной, называется квадратичной функцией , что позволяет нам значительно сжать предыдущее утверждение. Перемещение является квадратичной функцией времени при постоянном ускорении
Формулировки пропорциональности полезны, но не столь общие, как уравнения.Мы до сих пор не знаем, каковы константы пропорциональности для этой проблемы. Один из способов понять их — использовать алгебру.
Начнем с определения средней скорости.
Расширить ∆ с до с — с 0 и сжать ∆ т до т .
Найдите позицию.
с = с 0 + vt [a]
Чтобы продолжить, нам нужно прибегнуть к небольшому трюку, известному как теорема о средней скорости или правило Мертона .Я предпочитаю последнее, так как правило может применяться к любой величине, которая изменяется с одинаковой скоростью, а не только к скорости. Правило Мертона было впервые опубликовано в 1335 году в Мертон-колледже, Оксфорд, английским философом, математиком, логиком и калькулятором Уильямом Хейтсбери (1313–1372). Когда скорость изменения величины постоянна, ее среднее значение находится на полпути между ее конечным и начальным значениями.
v = ½ ( v + v 0 ) [4]
Подставьте первое уравнение движения [1] в это уравнение [4] и упростите его, исключив v .
v = ½ [( v 0 + при ) + v 0 ]
v = ½ (2 v 0 + при )
v = v 0 + ½ at [b]
Теперь замените [b] на [a], чтобы исключить v [vee bar].
с = с 0 + ( v 0 + ½ при ) t
И наконец, найдите s как функцию от t .
с = с 0 + v 0 т + ½ при 2 [2]
Это второе уравнение движения . Он записывается как полином — постоянный член ( s 0 ), за которым следует член первого порядка ( v 0 t ), за которым следует член второго порядка (½ при 2 ). ). Поскольку старший порядок равен 2, правильнее называть его квадратичным .
Символ s 0 [ess naught] часто рассматривается как начальная позиция . Символ s — это позиция на t позже. Если хотите, вы можете называть ее конечной позицией . Изменение положения (∆ s ) называется смещением или расстоянием (в зависимости от обстоятельств), и некоторые люди предпочитают писать второе уравнение движения таким образом.
∆ с = v 0 t + ½ при 2 [2]
скорость-позиция
Первые два уравнения движения описывают одну кинематическую переменную как функцию времени.По сути…
Скорость прямо пропорциональна времени при постоянном ускорении ( v ∝ t ).
Смещение пропорционально квадрату времени при постоянном ускорении (∆ с ∝ т 2 ).
Объединение этих двух утверждений приводит к третьему, не зависящему от времени. При замене должно быть очевидно, что…
Смещение пропорционально квадрату скорости при постоянном ускорении (∆ s ∝ v 2 ).
Это утверждение особенно важно для безопасности вождения. Когда вы вдвое увеличиваете скорость автомобиля, требуется в четыре раза больше расстояния, чтобы его остановить. Увеличьте скорость втрое, и вам понадобится в девять раз больше расстояния. Это хорошее практическое правило, которое следует запомнить.
Концептуальное введение сделано. Пришло время вывести формальное уравнение.
способ 1
Объедините первые два уравнения вместе таким образом, чтобы исключить время как переменную. Самый простой способ сделать это — начать с первого уравнения движения…
v = v 0 + at [1]
решить на время…
, а затем подставим его во второе уравнение движения…
с = с 0 + v 0 т + ½ при 2 [2]
как это…
с =
с 0 + в 0
⎛ ⎜ ⎝
в — в 0
⎞ ⎟ ⎠
+ ½ a
⎛ ⎜ ⎝
в — в 0
⎞ 2 ⎟ 90 840 ⎠
с — с 0 =
vv 0 — v 0 2
+
v 2 -2 vv 0 + v 0 2
2 a
2 a ( с — с 0 ) = 2 ( vv 0 — v 0 2 ) + ( v 2 — 2 vv 0 + v 0 2 )
2 a ( с — с 0 ) = v 2 — v 0 2
Возведите объект в квадрат скорости, и все готово.
v 2 = v 0 2 + 2 a ( s — s 0 ) [3]
Это третье уравнение движения . Еще раз, символ s 0 [ess naught] — это начальная позиция , а s — это позиция через некоторое время t позже. Если вы предпочитаете, вы можете написать уравнение, используя ∆ s — изменение в позиции , смещение или расстояние в зависимости от ситуации.
v 2 = v 0 2 + 2 a ∆ s [3]
метод 2
Более сложный способ вывести это уравнение — начать со второго уравнения движения в этой форме…
∆ с = v 0 t + ½ при 2 [2]
и решите ее на время. Это непростая работа, поскольку уравнение квадратично. Переставьте термины так…
½ при 2 + v 0 t — ∆ s = 0
и сравните его с общей формой квадратичного.
ось 2 + bx + c = 0
Решение этой задачи дает известное уравнение…
x =
— b ± √ ( b 2 — 4 ac )
2
Замените символы в общем уравнении эквивалентными символами из нашего преобразованного второго уравнения движения…
т =
— v 0 ± √ [ v 0 2 — 4 (½ a ) (∆ s )]
2 (½ a )
почисти немного…
т =
— в 0 ± √ ( в 0 2 -2 a ∆ с )
, а затем подставьте его обратно в первое уравнение движения.
v = v 0 + at [1]
v = v 0 + a
⎛ ⎜ ⎝
— в 0 ± √ ( в 0 2 -2 a ∆ с )
⎞ ⎟ ⎠
Материал отменяется, и мы получаем это…
v = ± √ ( v 0 2 + 2 a ∆ s )
Выровняйте обе стороны, и все готово.
v 2 = v 0 2 + 2 a ∆ s [3]
Это было не так уж плохо, не так ли?
исчисления выводов
Исчисление — это сложная математическая тема, но она значительно упрощает вывод двух из трех уравнений движения. По определению, ускорение — это первая производная скорости по времени. Возьмите операцию в этом определении и отмените ее. Вместо того, чтобы дифференцировать скорость, чтобы найти ускорение, интегрируйте ускорение, чтобы найти скорость.Это дает нам уравнение скорость-время. Если предположить, что ускорение постоянное, мы получим так называемое первое уравнение движения [1].
=
дв
=
a dt
=
v — v 0
=
при
в
=
v 0 + at [1]
Опять же, по определению, скорость — это первая производная положения по времени. Выполните эту операцию в обратном порядке. Вместо того, чтобы различать положение для определения скорости, интегрируйте скорость, чтобы найти положение. Это дает нам уравнение положения-времени для постоянного ускорения, также известное как второе уравнение движения [2].
в
=
DS
=
в dt
DS
=
( v 0 + at ) dt
=
т
⌠ ⌡
( v 0 + at ) dt
0
с — с 0
=
v 0 t + ½ при 2
с
=
с 0 + v 0 t + ½ при 2 [2]
В отличие от первого и второго уравнений движения, нет очевидного способа вывести третье уравнение движения (то, которое связывает скорость с положением) с помощью расчетов.Мы не можем просто перепроектировать это по определению. Нам нужно разыграть довольно изощренный трюк.
Первое уравнение движения связывает скорость со временем. По сути, мы вывели его из этой производной…
Второе уравнение движения связывает положение со временем. Это произошло от этой производной…
Третье уравнение движения связывает скорость с положением. По логике, это должно происходить от производной, которая выглядит так…
Но что это значит? Ну, ничего по определению, но, как и все количества, оно равно самому себе.Он также равен самому себе, умноженному на 1. Мы будем использовать специальную версию 1 ( dt dt ) и специальную версию алгебры (алгебра с бесконечно малыми). Посмотрите, что происходит, когда мы это делаем. Мы получаем одну производную, равную ускорению ( dv dt ), и другую производную, равную обратной скорости ( dt ds ).
дв
=
дв
1
DS
DS
дв
=
дв
дт
DS
DS
дт
дв
=
дв
дт
DS
дт
DS
дв
=
a
1
DS
в
Следующий шаг, разделение переменных.Соберите вместе похожие вещи и интегрируйте их. Вот что мы получаем при постоянном ускорении…
=
в дв
=
и DS
=
½ ( v 2 — v 0 2 )
=
a ( с — с 0 )
в 2
=
v 0 2 + 2 a ( с — с 0 ) [3]
Безусловно, умное решение, и оно было не так уж сложно, чем первые два варианта. Однако на самом деле это сработало только потому, что ускорение было постоянным — постоянным во времени и постоянным в пространстве. Если бы ускорение каким-либо образом менялось, этот метод был бы неудобно трудным. Мы вернемся к алгебре, чтобы спасти наше здравомыслие. Не то чтобы в этом что-то не так. Алгебра работает, а здравомыслие стоит сэкономить.
v =
в 0 + в
[1]
+
с =
с 0 + v 0 t + ½ при 2
[2]
=
v 2 =
в 0 2 + 2 a ( с — с 0 )
[3]
Кинематические уравнения и постоянное ускорение
В своих «Диалогах двух новых наук» Галилей вывел взаимосвязь между пройденным расстоянием и временем, когда шары катились по наклонной плоскости.Это часто называют законом падающих тел. Интересно, что в доказательстве Галилея вместо алгебры, которую мы представим здесь, использовалась классическая евклидова геометрия (которая была бы незнакома современному изучающему геометрию из учебников). Учащиеся продвинутого уровня могут получить те же уравнения, используя математический анализ.
Основа Закона падающих тел заключается в том, что по мере того, как мяч катится по рампе, он ускоряется. По мере увеличения его скорости увеличивается расстояние, которое он проходит за каждую единицу времени.Галилей определил это с помощью колокольчиков на спусковом крючке катящегося шарика.
Процитируем Галилея в переводе:
По сути, Галилей представил, что не только ускорение вниз по рампе из-за постоянной силы тяжести, но и что скорость увеличивается линейно с временем . Он представил, что положение увеличивается с квадратом времени, что часто называют Законом падающих тел. Последний пункт в этом отрывке, который он представил, заключается в том, что скорость увеличивается с квадратом расстояния вниз по рампе.
Основываясь на том, что вы уже узнали и что представил Галилей, у нас есть то, что мой учитель физики, Гленн Глейзер, любил называть пятью священными уравнениями кинематики для постоянного ускорения. В этих уравнениях v — скорость, x — положение, t — время и a — ускорение. Помните, что Δ означает изменение.
1. или Δx = v ср. Δt
2. или v f = v o + aΔt или Δv = aΔt
3.
4. Δx = v o Δt + ½ a Δt 2
5. v f 2 = v o 2 + 2aΔx
Первые два уравнения, которые мы видели ранее. Важно отметить, что в первом уравнении используется средняя скорость , тогда как во втором уравнении используется изменение между исходной и конечной скоростью . Связь между ними представлена в третьем уравнении, которое представляет собой просто закон средних чисел.Средняя скорость — это среднее значение исходной и конечной скорости.
Из этих трех основных определений мы можем вывести следующие два уравнения, используя либо геометрию, либо алгебру (или исчисление).
Используя алгебру, мы можем вывести уравнение №4.
Исходя из уравнения № 1
Δx = v ср. Δt
Затем мы подставляем определение средней скорости из уравнения №3.
Отсюда мы подставляем окончательную скорость, полученную в уравнении № 2
Затем мы распределяем член Δt и упрощаем, комбинируя члены v o .
Мы упрощаем оставшиеся два члена, чтобы получить
Стоит отметить, что происходит, когда исходная скорость v o, равна нулю. Это уравнение еще больше упрощается и становится
.
Если мы предположим, что исходная позиция и время равны нулю, мы можем дополнительно уменьшить это до
Используя геометрию, мы можем исследовать область под кривой графика зависимости скорости от времени для движения с постоянным ускорением.
Если мы посмотрим на область под кривой, мы можем разбить ее на прямоугольник и треугольник. Красный прямоугольник — это вклад исходной скорости объекта. Смещение из-за ускорения представлено зеленым треугольником. Треугольник имеет ширину Δt и высоту aΔt, которые мы знаем из уравнения №2. Член ½ происходит от формулы площади треугольника.
Мы также можем использовать исчисление для вывода этого уравнения путем интегрирования удвоенного ускорения по времени.
Пятое священное уравнение может быть получено аналогичными заменами, и его оставят как домашнее задание.
Теперь давайте рассмотрим несколько примеров задач: Численное решение задач.
Пример 1
По легенде, Галилей уронил мяч из Пизанской башни. Если башня имеет высоту 55,9 м и пренебрегает сопротивлением воздуха, сколько времени потребуется свинцовому мячу, чтобы достичь земли?
Гивенс: a = g ≈ 10 м / с 2
Δx = 55.9 м
Неизвестно: t = ???
Уравнение, связывающее эти переменные, — это священное уравнение 4 th .
Δx = v o Δt + ½ a Δt 2
Как упоминалось ранее, поскольку начальная скорость равна нулю, уравнение упрощается.
Δx = v o Δt + ½ a Δt 2 = ½ a Δt 2
Поскольку мы хотим изолировать переменную для времени, мы пересекаем умножение, чтобы переместить ½ и член ускорения на другую сторону.
Затем извлекаем квадратный корень из обеих частей.
Это дает выражение для времени. Обратите внимание, что я вставил несколько дополнительных скобок, которые могут вам не понадобиться.
При подключении номеров это довольно просто то, что мы называем «подключи и забей». Однако с агрегатами нужно быть осторожным. Вы, наверное, догадались, что время будет измеряться в секундах. Однако у вас должна быть возможность отменить фактические единицы, чтобы получить время в секундах.
Пример 2
Койот падает со скалы высотой 25 метров. Как быстро койот падает, когда ударяется о землю? Если проблема койота
Дано x = 25 м
a = g ≈ 10 м / с 2
Неизвестно: v = ???
Эту проблему можно решить несколькими способами. Можно было использовать комбинацию или Священные уравнения №2 и №4. Или вы можете напрямую использовать уравнение №5.
Использование v f 2 = v o 2 + 2aΔx
Это упрощается, поскольку исходная скорость v o, равна нулю.
Если извлечь квадратный корень из обеих частей уравнения
Обратите внимание, как вы извлекаете квадратный корень из единиц, чтобы получить м / с .
Мы оставим решение этой задачи с двумя уравнениями для домашней задачи.
Краткое изложение проблем построения графиков, наклона и площади под кривыми
Изучая графики положения, скорости и ускорения, вы сможете рисовать их как взаимозаменяемые.
Вчера в классе вы видели, что график объекта, ускоряющегося вниз по склону, выглядит следующим образом:
В этом примере мы используем программное обеспечение для анализа изображений.Это пример с мячом, катящимся с холма. Следует отметить, что график ускорения не показывает фактическую скорость, а показывает только то, как она меняется. Точно так же график скорости не дает вам фактического положения объекта, а только того, как он изменяется. Щелкнув по мячу и нажав кнопку трека, вы увидите график положения и скорости.
Здесь вы можете увидеть результаты построения графика движения.
20+ правил современного этикета, которые стоит знать воспитанным людям
Ребята, мы вкладываем душу в AdMe.ru. Cпасибо за то, что открываете эту
красоту. Спасибо за вдохновение и мурашки. Присоединяйтесь к нам в Facebook и ВКонтакте
Если вы думаете, что этикет распространяется только на то, какой вилкой правильно есть салат или как открывать дверь девушке, то вы сильно удивитесь, узнав, насколько глубоко и широко, а иногда даже философично это понятие.
Тренер по этикету и деловому общению Оксана Зарецкая поделилась с AdMe.ru основными советами, которые сейчас наиболее актуальны. Ведь, согласитесь, со временем этикет претерпевает весьма существенные изменения.
При разговоре
В этикете есть 2 главных правила. А из них вытекают уже все остальные.
Правило № 1: Следует стремиться никого не обидеть и не поставить в неловкое положение. Если обидели, извинитесь.
Правило № 2: Среда диктует этикет. Необходимо адаптироваться под ситуацию и окружение и говорить на языке собеседника.
При разговоре с человеком смотреть лучше на него, вне зависимости от степени знакомства. Поэтому существует правило на запрет солнцезащитных очков, кепок и шляп во время разговора. Должны быть видны глаза.
Считается дурным тоном использовать на людях интимные именования. Всех «крошек» и «пупсиков» придется оставить дома. В обществе принято обращаться по имени или в формате, в котором с человеком разговаривают другие участники общения.
Ошибки и замечания
Не обращать внимание на слабости и ошибки окружающих — важное правило этикета. Кто-то что-то уронил, неправильно поставил ударение, споткнулся, ошибся дверью или спряжением, не застегнул пуговицу — мы этого «не видим», не обсуждаем, не делаем замечание. Поправлять другого человека — это моветон.
Самым правильным в ситуации, когда кто-то допустил ошибку в постановке ударения, будет не заметить этого. Но когда у вас появится возможность произнести слово, произнесите его корректно.
Если замечания все-таки не избежать, сделайте это один на один. Отведите человека в сторону и, попросив прощения, обозначьте ситуацию в формате я-высказывания: «мне неудобно», «я чувствую себя неуютно», «я взволнована / обеспокоена». Добавьте слова вежливости: «могли бы вы», «я был бы признателен».
Вмешательства будут уместны, когда ситуация критическая или опасна для жизни. А еще когда человек проявляет хамство и мешает окружающим. А также в том случае, когда недочет можно исправить в тот же момент. То есть подсказать, что у девушки потекла тушь, будет нормой. Но говорить о неприятном запахе в такси будет некорректно, так как эту ситуацию водитель не сможет исправить в ту же минуту.
На работе
Сегодня деловой этикет настаивает на более взвешенном отношении к работе. Задерживаться после 18:00, возвращаться в офис на выходных и «быть доступным» во время отпуска стало дурным тоном. Как со стороны сотрудника, так и со стороны руководителя, требующего этого.
Надо научить себя отключаться и менять деятельность. По возможности отказывайтесь от деловых звонков и сообщений в мессенджерах после окончания рабочего дня и на выходных.
Даже если вы получили деловое сообщение в 9 вечера и это «не горит», отложите ответ до утра. Так вы продемонстрируете автору, что готовы к работе только в определенное время.
Отметьте в соцсетях и своей деловой почте свои рабочие часы. Для партнеров и клиентов это послужит прямым указанием, когда вы доступны по деловым вопросам.
Если в виде исключения вам приходится работать во время своего законного отпуска, нет нужды делать вид, что вам это нравится. Нормальным, с точки зрения этикета, будет упомянуть о компенсации.
На корпоративе
www.adme.ru
Основные правила этикета в обществе. Этикет в современном обществе
Правила этикета в обществе – это умение вести себя во всех ситуациях, в которых только может оказаться человек. В современном мире необычайно важно знать их, иметь хорошие манеры, чтобы быть довольной собой и другими, относиться ко всем людям уважительно, приветливо, доброжелательно, естественно. Чтобы любое, даже самое лучшее элитное общество охотно приняло вас в свои ряды.
Трактовка термина
Этикет в современном обществе — перечень общепринятых правил, которые касаются поведения человека по отношению к другим людям в определенных жизненных случаях.
Разделяют несколько основных видов таких правил.
Умение подать себя – правила формирования гардероба, внешний вид, уход за собой, физическая форма и осанка, походка, позы, жесты.
Речевой этикет – умение правильно говорить приветствия, комплименты, благодарности, подавать реплики; правила прощания, вежливость, манера речи.
Правила этикета в обществе – как вести себя в музее, на выставке, в театре, ресторане, суде, библиотеке, магазине, офисе и пр.
Деловой этикет – отношения с коллегами, начальством, хорошие манеры в бизнесе, умение вести деловые переговоры и т. д.
Умение подать себя
Хорошие манеры, правила этикета, умение быть любезным человеком — все это требует не только навыков, но и знаний в этих областях. Современный человек должен знать, как держаться в любых обстоятельствах, уметь вести себя соответственно, быть любезным, дружелюбным и уверенным в себе.
Этикет в одежде
Первое впечатление является самым сильным и запоминающимся, а кроме того, ум проявляется в выборе одежды по случаю. Чтобы произвести хорошее впечатление, недостаточно быть модно или дорого одетой. Если вы хотите нравиться окружающим, то должны считаться с ними и учитывать разные обстоятельства. Поэтому даже в формировании гардероба принято соблюдать правила этикета в обществе. Важно, чтобы одежда была красивой и шла вам, но гораздо важнее, чтобы все детали внешнего вида органично сочетались между собой, а сам он соответствовал времени, месту и обстановке. Днем не принято надевать вечерние наряды, а на работу — носить одежду для отдыха. Каждый раз, выбирая, что надеть, вы должны учитывать обстановку, соответствующий повод, время, место, не забывать про собственный возраст, особенности фигуры. Все, что на вас надето, всегда должно быть чистым, подшитым, застегнутым и отглаженным. Выходной наряд всегда должен быть в полной готовности. Формируя свой гардероб, помните, что в него должны входить обязательные вещи, такие как костюмы, строгие брюки и юбки, блузки и вечерняя одежда, а также домашние комплекты.
Уход за собой
Хорошие манеры предполагают обязательное соблюдение правил гигиены, чистоты одежды, правильное питание и здоровый образ жизни. Недопустимо появляться в обществе неухоженным. При этом важно следить за внешностью в комплексе, аккуратно убирая волосы, выходя в «свет». Это обязательные правила этикета и поведения для девушки, равно как и для мужчины.
Хорошие манеры поведения в обществе
Умение подать себя начинается с походки, осанки, жестов, поз, манеры садиться и сидеть. Правила этикета в обществе требуют красивой походки с прямой осанкой, когда руки нешироко двигаются в ритме шага, плечи выпрямлены, живот подтянут. Нельзя задирать голову высоко, но и не стоит ходить с опущенной головой. Не менее важны позы и жесты. Чтобы произвести хорошее впечатление, нужно вести себя просто и естественно. Дурным тоном считается манера вертеть что-либо в руках, накручивать на палец волосы, барабанить пальцами по столу, притопывать в такт музыке, трогать руками какие-либо части тела, теребить другого за одежду. Что касается вопроса о том, как правильно сидеть, то здесь важно знать всего два правила: не закидывать ногу на ногу и не разваливаться, раскинув ноги и руки в стороны.
Речевой этикет
Вежливые слова – особенные формулы, в которых зашифрован большой объем информации, как смысловой, так и эмоциональной. Необходимо знать их наизусть, уметь выбирать наиболее подходящие случаю и вовремя произносить их соответствующим тоном. Виртуозное, правильное владение этими словами и есть речевой этикет в современном обществе.
1. Приветствие
Выбирая форму приветствия, вкладывайте в слова достаточно смысла и чувства. Например, вы поступите не очень деликатно, сказав «добрый день» человеку, по лицу которого видно, что он чем-то расстроен. Или совсем недопустимо говорить начальнику «привет», за исключением случаев личной дружбы. Будьте внимательны к словам и людям – приветствуя их, называйте по имени или по имени-отчеству. Мужчины должны сопровождать приветствие друг друга рукопожатием. При встрече с дамой галантный кавалер целует ей руку, при этом он не должен подтягивать ее к себе, а обязан наклониться настолько, насколько женщина подала руку.
2. Обращение, представление
Какое из обращений предпочтительнее, приходится решать в каждом конкретном случае, в зависимости от той аудитории, к которой вы обращаетесь. К знакомым принято обращаться по имени или по имени и отчеству, второе считается проявлением большего уважения. В официальной обстановке, представляя кого бы то ни было, называйте имя и фамилию. А обращение по отчеству, например Ивановна, допустимо только в деревне, но никак не в светском обществе.
3. Просьбы
Слово «пожалуйста» действительно волшебное, оно обязательно должно звучать во всех просьбах. Поскольку просьба так или иначе обременяет того, к кому вы обращаетесь, в некоторых случаях стоит прибавить: «Если тебе не трудно», «Вас не затруднит?» Также уместно говорить: «Сделайте одолжение, будьте любезны, вы не могли бы» и пр.
4. Прощание
Перед тем как попрощаться, следует подготовить собеседника к расставанию: «Уже поздно», «К сожалению, мне пора». Затем принято выразить удовлетворение временем, проведенным вместе, например «Я рад, что мы встретились». Следующий этап прощания – слова благодарности. Иногда можно сказать комплимент хозяйке дома, попрощаться и сразу уходить, не задерживаясь.
Кроме того, правила этикета в обществе предполагают умение приглашать, извиняться, утешать, выражать соболезнование, благодарность. Каждая из этих форм обращения должна звучать естественно, искренне, исключая грубые и резкие фразы и словосочетания.
Столовый этикет
Красиво есть так же важно, как хорошо двигаться и говорить, но именно здесь нужно особенно соблюдать меру.
Не нужно пытаться специально приукрасить процесс еды, например, кушать очень маленькими кусочками, отставлять согнутые пальцы. Достаточно не открывать рот во время жевания, не разговаривать с набитым ртом, тщательно пережевывать пишу, прежде чем положить в рот очередную порцию.
Никогда не пейте, пока не проглотите еду, за исключением случая, когда вы неожиданно взяли в рот горячую пищу. Если видите, что еда горячая, не дуйте на неё, прежде чем начать есть.
Старайтесь есть и пить абсолютно бесшумно.
В обществе хлеб едят, не откусывая от целого куска, а отламывая от него кусочки.
Соль из открытой солонки, если в ней нет специальной ложечки, положено брать концом чистого ножа, насыпав ее после на край своей тарелки.
Кетчуп или горчицу в качестве приправы предлагают только в самой непринужденной обстановке.
Во время еды старайтесь как можно меньше испачкать свою тарелку, не перемешивайте и не размазывайте еду на ней.
Никогда, даже дома, не ешьте руками. Вилку принято держать в левой руке, а нож в правой. Если вы едите салат, то вилку можно взять правой рукой.
Если вы хотите попить или сделать перерыв в еде, то нужно оставлять вилку и нож в положении крест-накрест или «домиком».
Ложку всегда берут правой рукой, если вы едите из суповой миски, ложку после еды оставляют там, не выкладывая на стол.
По окончании еды и перед тем, как попить, принято пользоваться салфеткой.
Этикет: правила поведения в обществе и общественных местах
В общественных местах существуют некоторые специфические правила хорошего тона, соблюдать которые необычайно важно.
1. В музее, на выставке, вернисаже
Правила поведения в этих «храмах» искусства во всем мире одинаковы и чрезвычайно просты: ходите по залам тихо, разговаривайте приглушенным тоном, ничего не трогайте руками, не подходите слишком близко к картинам и экспонатам, чтобы не мешать другим посетителям.
2. В театре, филармонии, концертном зале
Современные правила хорошего тона несколько противоречивы. Раньше в такие общественные места дам должен был приглашать мужчина, сегодня считается вполне приличным, если девушка сама приглашает его на спектакль, концерт. И даже если именно она оплачивает билеты на двоих. Хорошо воспитанный мужчина должен исполнить роль галантного кавалера, всюду ухаживая за дамой. Важно приходить вовремя, спокойно раздеться, занять место, никому не мешая. Люди с безупречным воспитанием не должны ничего жевать во время просмотра.
3. В суде, церкви, поликлинике, библиотеке
Правила этикета и хорошего тона в обществе призывают вести себя в этих местах как можно тише и незаметней. Нельзя разговаривать, шуршать, жевать и ходить без особой необходимости. На обращения и вопросы следует отвечать вежливо и вполголоса.
В любом учреждении важно сохранять хорошие манеры, быть любезным, тактичным и вежливым. Главное, ваше пребывание не должно доставить дискомфорта ни одному из присутствующих.
Деловой этикет
Хорошие манеры на работе – обязательное условие для каждого работника. Какие моменты затрагивает деловой этикет? Легкие правила помогут разобраться в этом вопросе.
Соблюдение субординации с коллегами и начальством.
Своевременный приход на работу и быстрое выполнение своих обязанностей.
Вежливое общение как с коллегами, так и с посетителями.
Конфиденциальность в работе.
Соответствие одежды учреждению, в котором вы работаете.
Отсутствие личных тем в обсуждениях.
Поддержание порядка на своем рабочем месте.
Умение общаться по телефону.
Правила делового этикета в обществе помогают достичь целей, назначенных в бизнесе. Благодаря хорошим манерам, можно продвигаться по служебной лестнице и во всем быть успешной самореализовавшейся личностью.
Чтобы быть приятным человеком в любой ситуации, чтобы с вами хотели иметь дело, нужно в совершенстве знать законы поведения в обществе. Они помогут не только достичь любых целей, но и стать уверенным в себе и счастливым человеком.
fb.ru
Доклад-сообщение Правила речевого этикета 7 класс кратко
Независимо от того, с какой ситуацией сталкивается человек в современном мире, он должен помнить о нормах, применимых в тот или иной момент. Правила могут касаться и внешней формы поведения человека, его мимики, жестикуляции и речи. Повседневная речь человека дает точную характеристику людям – насколько они дисциплинированные, искренние, ответственные. Они являются приметой вежливости. Это связано с тем, что детям с ранних лет прививают понятие о правилах общения. Взрослый, в силу своей образованности и воспитания, в соответствии со своим культурным уровнем, проявляющимся через взаимоотношения с людьми, придерживается правил речевого этикета. Находясь в обществе, человек в основном неосознанно применяет те или иные выражения, являющиеся показателем норм, которые имеют место в определенных ситуациях речевого этикета. Однако четкое знание этого раздела, знакомство с системой правил помогают получить высокую репутацию в обществе, заслужить уважение окружающих, порой – быть незаменимым.
Вся система этих правил условно делится на группы. Выражения, с которых должно начинаться, согласно правилам речевого этикета, утро каждого человека, представляют собой фразы: «Доброе утро!», «Доброго дня!». Момент прощания сопровождается известными всем словами: «До свидания», «Всего доброго», «Пока». При этом на данном этапе задействованы жесты такие, как рукопожатие, кивок. Также к наиболее употребляемым относятся просьбы: «Прошу вас», «Будьте добры»; слова благодарности: «Спасибо», «Благодарю»; извинения: «Простите», «Извините».
При этом существуют определенные грани между выражениями, которые относят их к либо к официальной речи, когда речь идет о деловых переговорах, либо близки к разговорной (в домашней обстановке, при встрече с друзьями). В соответствии с этим необходимо учитывать интонацию, особую лексику, тактичные формы отказа. Учитывая эти особенности, можно смело добиваться цели на работе, в школе или в повседневной домашней обстановке.
Следование, а иногда и специальное обучение этим правилам, чрезвычайно важно. Оно облегчает общение с окружающими людьми, помогает установить желаемые контакты с ними, в трудных случаях – облегчает понимание действий. Доверительная атмосфера делает диалог безопасным в плане возможного появления недоверия. Выполняя эти требования, человек в силах взять ситуацию под контроль, управлять ею, не теряя дружеских отношений.
Чтобы выстроить эти отношения, необходимо избегать оскорбительных фраз, несмотря на сферу общения. В противном случае обида собеседника приведет к нежелательному результату. Умение выслушать – важнейший показатель знаний правил речевого этикета. Оно подчеркивает уважение друг к другу. Использование допустимой в конкретной обстановке мимики и жестов, а также громкости голоса имеет большое значение для общего расположения к себе людей. Линия поведения в подобных ситуациях должна быть продуманной и оценена заранее.
Умение преподносить комплименты – важный шаг в подаче себя коллективу. Проявление неискренности через набор фраз может дать обратный результат.
Этикет телефонных переговоров в современном мире электроники и техники должен строго соответствовать выбору собеседника, сфере, в которой происходит общение. Хотя в нем отсутствует жестикуляция, однако существует немало строгих правил общения для данного вида связи (приветливость, выбор голоса, тона, интонации, умение сдерживаться, тактичность).
Таким образом, успех во взаимоотношениях с окружающими людьми зависит от желания человека. Имея необходимый багаж знаний, опыта, некоторой накопленной со временем мудрости и наблюдений в области речевого общения, можно легко добиться цели. Если на первое место в ходе беседы становится доброжелательность и выдержанность, а жесты являются уместными, соответствуя обстановке, то симпатия собеседника обеспечена. Постигая эту истину день за днем, человек делает свое окружение подобным себе, выбирая собеседников в зависимости от пола, национальности, уровня образованности и культуры. Добившись применения речевых норм на практике, можно с уверенностью говорить о собственных достоинствах.
Доклад Правила речевого этикета сообщение
Оказывается, при помощи речи любого человека можно понять, насколько он образован, дисциплинирован и степень его ответственности для решения той или иной проблемы. Кроме этого при помощи речи можно определить, как ваш собеседник относится не только к окружающим или своим родственникам, но и к себе. Именно поэтому если вы хотите стать успешным человеком, то в этом случае нужно постоянно регулировать, контролировать и развивать свою речь. Обычно правильному речевому этикету каждого человека учат родители еще в детстве, и самое главное, не забыть его, когда вырастите.
Этикетом называется нормы и правила, которые усваиваются при общении друг с другом. Именно поэтому если вы хотите начать и дальше продолжить хорошие отношения с другим человеком, то следует следовать не только нормам, но и правилам. Этикетных формул существует очень много и для каждой из них имеются свои корни, функции или варианты решения.
Как, наверное, каждый знает о том, что к взрослому человеку нужно обязательно обращаться только на Вы. А вот со своими лучшими друзьями или родственниками можно обращаться на Ты. Близкому человеку вы можете сказать при встрече «Привет», а вот незнакомому человеку в этом случае нужно говорить «Здравствуйте».
Некоторые люди для того чтобы объяснить ситуацию начинают размахивать руками и очень сильно кричать или перебивать своего собеседника. Это не признаки хорошего тона. Кроме этого не нужно использовать при общении некультурные, бранные или матерные слова.
Правила общения меняются постоянно, старо устаревает и уходит в историю, а новые слова входят в нашу жизнь и становятся знаменитыми, популярными и употребляемыми в бытовой речи. Раньше было модно обращаться к вышестоящему сотруднику «Ваше величество», а вот сейчас так никто ни к кому не обращается.
Кроме этого некоторые правила этикета являются популярными в одном государстве и если их не употреблять, то могут привлечь к уголовной ответственности. А вот если они приехали в гости в другое государство, и начинают употреблять эти же слова, то их могут наоборот оштрафовать или выгнать из страны.
Правила речевого этикета 7 класс
Все люди любят слушать красивую, богатую речь. Именно по ней зачастую складывается первое впечатление о человеке, ведь на то, как мы говорим, всегда обращают большое внимание в обществе. Когда в каждом предложении слышится слово – паразит, ругательство и другие, негативно влияющие на культуру и язык слова, о даже самом прекрасном человеке автоматически складывается негативное впечатление, избавиться от которого будет совсем непросто.
В культуре речевого этикета существует достаточно большое количество простых правил, которым непременно необходимо следовать, чтобы считаться интеллектуально развитой, грамочной личностью.
В первую очередь, необходимо искоренить из речи слова – паразиты. Это те слова, которые не вносят никакого смыслового оттенка в предложение, и, как правило, используются человеком неосознанно, в процессе формулирования мыслей. К ним относятся слова: «ну, короче, как бы, это самое, ну как сказать» и многие другие. Иногда их называют словесным «мусором» потому, что их наличие в речи не приносит отнюдь никакой пользы.
Кроме того, необходимо обязательно использовать вежливые слова. К ним относятся слова, обозначающие приветствия, просьбы и благодарности: «здравствуйте, всего доброго, пожалуйста, спасибо» и прочие. Мало кто знает, что при задавании вопросов считается ошибкой, если вопрос начинается с отрицательной частицы: «Не подскажите? Не могли бы Вы помочь?», так как наталкивает оппонента на отказ.
Речевой этикет предполагает исключительно вежливое общение, с недопущением сквернословия, использования бранной лексики или чрезмерно эмоционально-окрашенных слов. Во время диалога важно проявлять уважение к своему собеседнику, не перебивать его, не дослушав до конца, толерантно относиться к чужому мнению. Любые противоречивые ситуации следует решать исключительно методом конструктивного диалога, в ходе которого каждая из сторон сможет спокойно высказать свою точку зрения, после чего будет найден определенный компромисс, если того требует ситуация. Ни в коем случае не следует переходить на личности, позволять себе оскорблять человека, говоря о нем нелицеприятные вещи. Даже если подобным образом поступает собеседник, напротив, следует не поддаваться на провокации, не повышать тон, только таким образом, можно дать понять, что Вы, как воспитанная личность, поступаете выше него.
Когда речь идет о монологическом выступлении человека, необходимо сохранять спокойствие, быть уверенным в себе и собственных силах, подкреплять все свои доводы логичными аргументами, делать паузы между высказываниями, там, где это уместно, допустима жестикуляция.
Таким образом, очевидно, что соблюдение правил речевого этикета играет невероятно важную роль. Так как все люди живут в социуме, где ежедневно вступают в различные коммуникации, важно придерживаться общекультурных правил, соблюдать установленные нормы – в поведении, поступках, словах. Помните, речь – это визитная карточка каждого человека, и этим нужно пользоваться!
7 класс кратко в русском языке
Правила речевого этикета
Популярные доклады
Сообщение-доклад Великая Китайская стена 2, 3, 4, 5 класс
Великая Китайская Стена является современным чудом света. Ее строили на протяжении более 2700 лет и ее длина составляет более 8850 км. Высота стены достигает на некоторых участках 9 м, а ширина – 6,5м. Главной функцией ее создания являлась защита
Доклад на тему Природа Московской области сообщение
Природа Московской области представлена разнообразием флоры и фауны: хвойные леса, прекрасные озера, широкие поля, богатый растительный и животный мир. На территории области находятся национальный парк «Лосиный остров», заповедник «Завидово»
Доклад про Клопов 7 класс сообщение
Клопы́ — относятся к типу членистоногие, представители класса насекомые, из отряда полужесткокрылых, одни из самых многочисленных насекомых, около 40000 тысяч видов, более 50 семейств.
more-dokladov.ru
Доклад Деловой этикет сообщение
Это такие правила поведения, которые должны соблюдать люди участвующее в деловых встречах. От выполнений этих правил будет завесить карьера и статус человека.
Если в обычном этикете при нарушении правил человек покажется невоспитанном, то в деловом этикете может полностью разрушится карьера и бизнес человека.
Многие считают, что этикет это вежливость и воспитанность. Как в любом этикете существуют принципы. Для начала это то, что к любому человеку, с которым вы в деловых отношениях относится вежливо и с уважением. Неважно как он относится к вам главное быть со всеми одинаково вежливыми. Правила делового этикета одинаковы как для мужчин, так и для женщин. Если вы хотите чтобы к вам относились хорошо, то и вы должны относиться так же. Всегда общайтесь культурно и вежливыми словами. Никогда не приходите на бизнес встречи под действием алкоголя и наркотических веществ.
Как вести себя, чтобы не нарушать деловой этикет.
Никогда не опаздывайте. Когда вы заставляете людей ждать, это выглядит некрасиво.
Не нужно слишком много разговаривать и отходить от темы встречи. Секреты компаний надо всегда хранить.
Нужно думать не только о себе, но и о других людях. Важно учитывать чужое мнение и всегда спрашивать все ли нравится клиентам или коллегам.
Важно понимать, что деловой этикет это еще и опрятный внешний вид. Нужно соблюдать деловой стиль и не надевать спортивную, яркую, повседневную одежду.
Правила беседы. Нужно понимать, что при деловых встречах нужно меньше говорить и больше слушать. Дайте своему собеседнику высказать все мысли по поводу той или иной ситуации. Каждый раз выключайте телефон, чтобы он не отвлекал звонками и сообщениями.
Доклад №2
Во все времена система ценностей в жизни человека играла главную роль. Благодаря им на протяжении многих лет складывались определенные требования в разных сферах, которые привели к понятию этика. Этика представляет собой целый ряд принципов, на которые должен равняться каждый человек в определенной ситуации.
Способность человека придерживаться правил делового этикета важна потому, что люди, которые вращаются в деловой сфере, обязаны иметь представление о своем внешнем виде, речи, манере общения, чтобы не терять деловых контактов.
Любое требование в области делового этикета касается взаимоуважения обеих сторон. Без этого не может быть продолжения общения. Кроме этого, существующие принципы распространяются на всех без исключения, несмотря на возрастные различия. Для этого вида этикета характерна официальность, поэтому большинство встреч включают фиксирование докладов, диалогов и т. д. с помощью протокола. При этом существует жесткая своеобразная дисциплина в поведении людей, проведении мероприятий, особенностях речи. Важными понятиями становятся строгость, подчинение, порядок. Немаловажное значение имеет одежда участников делового этикета, где заранее продумываются все детали, придерживающиеся строгости.
В деловой сфере существуют определенные темы, за границы которых нежелательно заходить. Личные интересы должны оставаться в тени. В противном случае официальность и порядок уходят на задний план. Стираются грани между повседневностью и рабочей обстановкой.
Умение вести беседу в сфере деловых отношений предполагает обязательное следование нормам официально-делового стиля. Это требует строгость в изложении мыслей, краткости и использование шаблонов.
Участие в деловых отношениях предполагает постоянный контакт с образованными людьми и серьезными документами. Чтобы соответствовать этому уровню, необходимы знания в ведении деловой переписки, определенных законов, связанных с этой деятельностью. Дисциплина действий требует определенного уровня добропорядочности, обязательной пунктуальности, повышенного внимания друг к другу, присутствие позитивного настроя. Особое место имеют умение хранить в тайне информацию, которая не требует разглашения, а также сдержанность при прослушивании критики в свой адрес.
Таким образом, чтобы добиться целей и успеха в определенном виде деятельности, необходимо соответствовать тем требованиям, в которых нуждается сфера деловой этики. Приобщение к культуре речи, знание моделей поведения в разных ситуациях, применение на практике требований официального стиля помогут человеку, участвующему в общественной жизни, легко следовать принципам деловой этике.
Деловой этикет
Популярные доклады
Доклад-сообщение Моллюски 2, 3, 5, 7 класс окружающий мир, биология
Древние жители морей и в частности планеты, зародились примерно 500 млн. лет назад. Большинство моллюсков имеют раковины. Ученые установили, что предшественниками моллюсков могли быть плоские черви. Эти создания имеют огромное количество видов,
Доклад на тему город Хабаровск
В 1858 году, как военный пост, был заложен город Хабаровск, его название происходит от русского землепроходца Е.П. Хабарова. Город растянулся по центру Дальнего Востока. Город имеет границу, по реке Амур с Китаем.
Доклад Песок полезное ископаемое 3, 4 класс сообщение
Песок – это ископаемое, представляющее собой мелкие зерна горных пород, которые накапливаются и формируют песчинки. Удары волн, выветривание, извержения вулканов и землетрясения разрушают горные породы и способствуют образованию песка.
more-dokladov.ru
Речевой этикет — виды, функции, основные правила
Этикет – это не только нормы поведения в обществе, но и умение правильно изъясняться. Это красота речи и ее содержание, а также использование фраз в зависимости от ситуации.
Речевой этикет — набор правил (гласных и негласных), благодаря которому в обществе поддерживаются социальные институты и устанавливается иерархия. В зависимости от культуры и социального класса правила речевого этикета могут значительно меняться.
Знание речевого этикета позволяет человеку успешно взаимодействовать с другими людьми, расти и развиваться в личностном и профессиональном плане.
Взаимосвязь культуры и речи
Культурный человек выделяется из общей массы манерами поведения, учтивостью, осведомленностью и коммуникабельностью. Подобная личность умеет вести себя в обществе, легко идет на контакт и может поддержать беседу.
Речь культурного человека отличается смысловой точностью, грамматической правильностью, выразительностью, богатством и разносторонностью словарного запаса и логической стройностью.
Такая речь называется нормированной – в своей устной форме она отвечает существующим в настоящее время нормам произношения, а в письменной форме – правилам пунктуации и орфографии.
Взаимосвязь культуры и речи здесь очевидна. Человек, который не имеет представление о морально-этических нормах, не сможет соблюдать речевой этикет, по следующим причинам:
отсутствие какого-либо образования и грамотности письма;
узкий кругозор;
отсутствие коммуникативного навыка;
обилие «сорных» слов в речи;
использование ненормативной лексики.
Важно! В некоторых случаях знание этикета не гарантирует достойного общения. Иногда это вопрос личностных качеств собеседника.
Формирование культуры общения
Общаться можно очень по-разному. В стенах кафедры государственного университета и, например, общественной столовой, звучит кардинально разная лексика, но правила речевого этикета в целом соблюдаются одни и те же.
Это происходит потому, что формирование культуры общения начинается ещё с младенчества. Дети, находящиеся в разных условиях, получают разное качество обучения поведению в обществе, но согласно одинаковым принципам (исключая маргинальные прослойки).
В минимальные стандарты культуры общения входит умение держать словесную дистанцию, отказываться от оскорблений и обсуждения недостатков вслух, недопустимость грубости и агрессии.
Чтобы успешно функционировать в обществе, молодой член общества должен научиться лояльности и минимальному уважению к окружающим.
Так как человечество больше не в племенном строе, уважение и доброжелательность высказывается с помощью речи и её выражениям — интонации, словам, жестикуляции.
Формирование культуры общения начинается с раннего возраста. Наряду с правилами поведения ребенку внушают и постулаты речевого этикета. Прямое и косвенное влияние на формирование речевой культуры оказывают:
семья;
ближайшее окружение;
учебное заведение.
Первый навык общения ребенок получает в семье. Едва начав говорить, он начинает копировать манеру речи домочадцев, используя при этом те же слова и интонации — речь ребенка становится отражением речи родителей и их задача донести до ребенка основы культуры общения.
В семьях, где воспитанию детей уделяется много внимания, дети с раннего возраста знают «волшебные слова» и их значение.
На втором этапе в процесс освоения речевых правил вмешиваются окружающие:
соседи;
случайные люди на улице;
друзья и их родители.
Круг общения ребенка становится шире, в речи появляются новые слова, меняется манера разговора. И то, какой она будет, теперь зависит не только от родителей.
Если ребенок проводит время среди воспитанных, культурных людей, значит, его речь станет богаче и ярче, но если окружающие незнакомы с культурой общения и «сорят» ненормативной лексикой, то ребенок непременно переймет некоторые обороты.
Детский сад, школа и другие учебные заведения учат читать и писать без орфографических и пунктуационных ошибок, а также правильно выражать мысли в устной и письменной форме.
Причем, необходимые знания ребенок получает из уроков русского языка и литературы, но и из других дисциплин. Весь учебный процесс направлен на формирование речевого этикета, а целью являются следующие моменты:
выработать общительность и общественную активность;
установить коммуникативные взаимоотношения с окружающими;
повысить академическую успеваемость
разить быструю адаптацию к разнообразной деятельности.
Что такое речевой этикет?
Речевой этикет – это совокупность требований к содержанию, характеру, форме, порядку и уместности высказываний в той или иной ситуации.
Это определенные правила речевого поведения, система специфичных стереотипных, устойчивых формул общения, которые приняты обществом для взаимного контакта собеседников, его поддержания и прерывания в избранной тональности.
Речевой этикет предполагает использование определенных слов и выражений в различных ситуациях:
во время приветствия;
в момент прощания;
при просьбе;
во время обращения;
в момент извинения.
Нужные слова и фразы произносятся с определенной интонацией, что в совокупности с изречениями и характеризует вежливую речь.
Владение культурой речи помогает в становлении личности, завоевании авторитета, доверия и уважения. Соблюдая речевой этикет, человек чувствует себя уверенно и непринужденно в любой ситуации, а также избегает насмешек и неловкости в незнакомой обстановке.
Это совокупность правил, уникальная для разных рас и социальных групп в некоторых аспектах. Большинство правил речевого этикета считается негласным и в норме воспитывается в детях вместе со всеми прочими социальными навыками.
Например, нет нужды в объяснении причин, по которым нельзя повышать голос на другого человека — это нарушение личного пространства и грубость.
Так же очевидно то, что не вежлива фамильярность с человеком, более высоким по социальному статусу или просто не знакомым.
История возникновения речевого этикета берет истоки из иерархических правил, где старший автоматически возвышался над младшим, женщин выделяли в отдельную социальную группу, а разрыв между социальными классами был невероятно огромным.
Большинство правил речевого этикета человечество сохранило неизменным или немного изменённым.
Основные правила речевого этикета
Речевой этикет предписывает личности определенные нормы общения, которые бывают обязательными и носящими характер рекомендаций.
К обязательным относятся следующие правила речи:
соблюдение правил и литературных норм в разговоре;
отсутствие ненормативной лексики;
недопущение бестактности, грубости и неуважения;
соблюдение обязательных этапов речи – начало разговора, основная часть беседы и заключение;
отсутствие ошибок и искажения терминологии.
Рекомендации речевого этикета предписывают:
говорить по существу, избегая пустых, бессмысленных слов.
вести беседу, учитывая уровень развития собеседника – изъясняться понятно для него;
не перебивать оппонента, выслушивать полностью;
быть вежливым и тактичным;
не переходить на личности во время спора;
соблюдать спокойную тональность.
Так как полностью структуризировать такое большое понятие не удастся — его принципами пользуется слишком много культур и социальных групп, есть только основные правила, приемлемые для большинства современных сообществ:
Ровная, нейтральная интонация. Повышение голоса и его понижение — это отклонение от нормы в стандартной беседе. Собеседники должны хорошо слышать друг друга, но окружающие, если они есть, не должны испытывать от чужой беседы никаких неудобств.
Приветствие и прощание. Каждая беседа обязательно должна начинаться с приветствия (его тип будет зависеть от ситуации) и прощания.
Представление, если в беседе более двух человек и кто-то с кем-то не знаком. Очень невежливо начинать разговор с другими, не представившись. Тот, кто приводит в компанию новое лицо, обязан его представить. Если в диалоге между несколькими людьми нет знакомых, правило строго не соблюдается.
Главные принципы — спокойствие, исключение конфликтных ситуаций и доброжелательная (нейтральная) атмосфера. При деловой беседе или любой другой формальной встрече ярко выражать свои эмоции и отношение к окружающим строго не рекомендуется.
Виды речевого этикета
Речь – это основной механизм вербальной коммуникации. Вербальное общение бывает внутренним, когда слова произносятся про себя, и внешне направленным – устным (диалог и монолог) и письменным.
Устная речь строится в виде диалога или монолога. В диалоге люди обмениваются между собой информацией, эмоциями или переживаниями. Монолог исходит от одного лица, но направлен на аудиторию или на себя.
Разговорная этика проявляется менее формально, нежели письменная. Здесь допускаются пропуски слов, замена фраз действием или жестом.
Письменная форма этики ограничена строгими рамками – стилистикой, правилами орфографии и пунктуации.
Так как это широкое понятие, нигде нет единого речевого этикета, идеально подходящего под все социальные требования. Конкретные люди или социальные группы видоизменяют правила под свои потребности, не изменяя главных принципов — так рождается классификация речевого этикета по видам:
Официальный или деловой. Это тот этикет, который обычно подразумевается под этим словом обывателем. Применяется на мероприятиях, где гости не знакомы друг с другом, на выставках, в сфере обслуживания, на деловых переговорах.
Повседневный. Наиболее простой для освоения и распространённый вид. Применение правил повседневного этикета не требует усилий, воспитанный и интегрированный в общество человек соблюдает большинство правил и норм речевого этикета в процессе общения автоматически. Применим в любой ситуации, где не подходит официальный этикет или более редкие формы речевого этикета.
Также под нестандартные ситуации, с которыми большинство людей не сталкивается, есть уникальные единицы речевого этикета этикета.
Например, религиозный — он изучается внутри духовенства конфессий или просто в среде верующих и в светском обществе практически не применим. То же можно сказать относительно дипломатического этикета и военного.
В целом, речевое общение классифицируется по содержанию и бывает:
материальным — обмен продуктами деятельности;
когнитивным (познавательным) – обмен данными, опытом и знаниями;
кондиционным (эмоциональным) – обмен настроением;
мотивационным – обмен намерениями;
деятельностным – обмен умениями, вследствие совместной деятельности.
Виды речевого этикета подразделяются по техникам взаимодействия и задачам.
Контакт масок. Это формальная коммуникация, без стремления узнать характер оппонента.
Светское общение. Эта форма словесной связи беспредметна, так как люди в такие моменты разговаривают на общие темы, о чем положено говорить в этой ситуации.
Формально-ролевой вид. Здесь важен регламент и содержание общения, а значение имеет социальный статус собеседника и его положение в обществе.
Деловое общение. Это взаимодействие с целью обмена данными и сообщениями, которые требуются для достижения нужного результата.
Межличностное общение. Этот вид речевого этикета еще называется интимно личностной коммуникацией, потому как заключается в раскрытии глубинных личностных качеств собеседника.
Манипулятивное общение. Это общение направлено на получение пользы от оппонента.
Важно! Любая форма разговора подчиняется определенному регламенту, соблюдать который нужно неукоснительно.
Функции речевого этикета
Речевой этикет имеет определенные функции, которые очень важны для человека.
Установление контакта. Речевой этикет привлекает внимание собеседника, побуждает его к контакту и возможному знакомству.
Поддержание контакта. В этом случае этическое общение способствует сохранению контакта без углубления в какую-либо тему разговора. Оно необходимо для составления впечатления о собеседнике и поддержания приятельской связи.
Демонстрация уважения и позитива. В некоторой степени это основная функция речевого этикета, которая осуществляется словами приветствия и прощания, извинения, сочувствия, просьбы и т.д.
Регуляция поведения. Соблюдение норм речи делает поведение людей предсказуемым и понятным для окружающих, а также проясняет социальную роль каждого из собеседников и определяет порядок действий в той или иной ситуации.
Профилактика конфликтов. Речевой этикет способствует нормальному общению между людьми. Своевременное извинение и вежливость помогают избегать острых углов в разговоре, а если конфликт уже начался – выйти из него с наименьшими потерями.
Важно! Этикетное общение – это обязательное условие беседы с окружающими, которое гарантирует нормальные отношения между людьми. Оно наделяет человека положительными качествами и облегчает взаимодействие с обществом.
Основная функция — установление положительного контакта с другим человеком или группой. Изменения в русском речевом этикете последних лет — всего лишь отголоски ритуалов, созданных древними людьми в качестве универсальной константы общения.
Очень многие их части можно отследить и сейчас, например, в рукопожатиях, поклонах у азиатских национальностей, и улыбках.
Все эти незначительные на первый взгляд мини-ритуалы сопровождают человечество веками. Они помогают показать на сознательном и не сознательном уровнях, что собеседник уважаем и отношение к нему будет хорошим.
Этикетные нормы — это универсальный язык, на котором можно договориться с каждым.
Языковые и поведенческие средства
Речь — это в основном слова и другие звуки, конечно, но есть и другие средства выражения. Например, жесты и положение в пространстве относительно своего собеседника.
Всё это тоже очень важно и имеет значение как со светской стороны, так и в плане национальных особенностей, которые тоже учитываются.
Ярчайшим примером поведенческого средства можно считать жестикуляцию. Это совершенно нормальное явление — жесты используются человеком как дополняющие речь «усилители».
С помощью них выражаются эмоции, подаются сверхбыстрые сигналы. Насчёт жестикуляции есть достаточно жёсткие правила, в основном они заключаются в её сдерживании.
Нет ничего плохого в том, чтобы показать собеседнику ладонью на предмет разговора или жестом пригласить пройти в помещение, но размахивать руками и сокращать дистанцию с человеком без его согласия недопустимо.
Языковые и поведенческие средства неразрывно связаны, но первые существуют без вторых, а наоборот — нет.
В речевом этикете первыми помощниками являются языковые и поведенческие средства. К ним относятся:
умеренная жестикуляция и мимика;
дистанция общения;
выраженная доброжелательность и сдержанная эмоциональность;
демонстрация интереса;
избегание спорных ситуаций;
некатегоричность собственных высказываний;
исключение неодобрения;
исключение излишнего интереса к личным подробностям;
участие в общей беседе;
краткость и равномерность общения со всеми;
минимум информации о себе;
обсуждение нейтральных тем – дети, животные, погода, путешествия;
помощь собеседнику в щекотливой ситуации;
выражение несогласия молчанием, вопросом или переходом на другую тему;
умеренное использование юмора;
запрет на сарказм;
исключение грубых и просторечных выражений;
положительное настроение;
соблюдение временных рамок и периодичности общения.
Формулы речевого этикета
На любом этапе общение сопровождается формулами речевого этикета – штампами и устойчивыми выражениями.
Это слова вежливости, которые предусмотрены на все случаи жизни:
слова приветствия и прощания – «здравствуйте», «приветствую вас», «до встречи», «до свидания»;
извиняющие фразы – «извините», «прошу меня простить», «простите за…»;
обращение – «могу я к вам обратиться?»;
слова сочувствия – «соболезную», «искренне сочувствую»;
просящие фразы – «будьте так добры, передайте…»;
пригласительные слова – «буду рад вас видеть»;
комплименты и поощрения – «вы замечательный специалист»;
благодарность – «от всего сердца благодарю», «спасибо», «я вам очень признателен».
Данные формулы подсказывают как себя вести в какой-либо ситуации и облегчают общение.
Речевой этикет и деловое общение
Деловое общение после повседневного является самым распространённым. Это логично — растёт уровень среднего предпринимательства, всё больше людей заняты на творческих профессиях или предпочитают работать на себя.
Придерживаться стандартных правил повседневного этикета на деловой встрече допустимо, но получить таким образом уважение от собеседника удастся только в том случае, если он сам придерживается подобного подхода в делах.
ВАЖНО! В зависимости от разных жизненных ситуаций деловой этикет тоже можно разделить на условные группы.
Правила для удачного официального общения
Главное — никакой фамильярности. Флирт между деловыми партнёрами также исключается. Собеседники должны найти идеальный баланс между вежливой отстранённостью и вежливой вовлечённостью. Первое не должно перейти в заносчивость, второе — в навязчивость.
Не стоит придерживаться картинного официоза. На деловой встрече вполне могут звучать уместные шутки и разговоры на отвлечённые темы. Переход на личное — табу, это грубо и может обидеть собеседника.
Пунктуальность, обязательность и честность. При создании первого впечатления не существует мелочей — не стоит опаздывать, грубить персоналу.
Деловое общение отличается тем, что в нем отсутствуют фрагменты, намекающие на личные темы. Это общение по существу – вежливое, учтивое и беспристрастное, но вместе с тем располагающее. Оно направлено на достижение взаимопонимания и контакт.
Официальное общение предусматривает следующие правила:
манеры и речь в соответствии с конкретной ситуацией;
предельная ясность речи – четкое произношение, ясность изложения;
достоверность информации;
корректность;
умеренность;
внимательность;
соблюдение дистанции.
Этапы делового общения
Как и любое общение, деловая беседа разбита на этапы:
приветствие – первым приветственное слово произносит младший по возрасту или рангу;
диалог, с соблюдением канонов и вежливости;
разрешение спорных ситуаций – умение обходить острые углы, конструктивный диалог;
повседневное взаимодействие – решение ежедневных вопросов;
невербальное общение – внимательность и радушие, выраженные в жестах и мимике;
прощание – заключительный этап общения, от которого зависит обоюдное впечатление.
Принципы речевого делового этикета
Соблюдение принципов делового общения помогает налаживать и устанавливать длительные партнерские отношения. К ним причисляются:
субординация;
позитивный образ и доверие;
внимательность к мнению оппонента;
учтивость;
ситуативность;
ориентирование на согласованный регламент.
Этикет деловых разговоров по телефону
Телефонные переговоры тоже имеют свой регламент:
начинаются они с приветствия и представления с названием организации и должности говорящего;
разговор должен быть лаконичным, по существу;
необходимо соблюдать последовательность беседы;
переговоры ведут вежливо, неторопливо, спокойным голосом;
дикция должна быть четкой;
после разговора требуется произнести слова прощания.
Важно! Прежде чем начать деловые переговоры, лучше написать суть вопроса на бумаге, чтобы в ходе разговора не перескакивать с одного момента на другой.
Речевой этикет разных социальных групп
Речевой этикет устанавливается внутри каждой социальной группы. Его особенности складываются в зависимости от следующих аспектов:
возраста;
половой принадлежности;
образования;
уровня воспитания;
профессионального направления;
уровня достатка;
иерархической принадлежности.
Стремление овладеть речевым этикетом – это залог развития личности и показатель воспитания.
Соблюдение норм и правил речи повышает культуру человека и общества в целом. Именно поэтому образовательный процесс уделяет этому вопросу максимум внимания.
logoprav.ru
Доклад-сообщение «Речевой этикет» | СПАДИЛО.РУ 👈
Хорошие манеры – это отличительная черта умных людей. Но какие манеры хорошие, а какие плохие? Речевой этикет рассказывает о хороших манерах в речи, которые помогут уверенно общаться с людьми.
Речевой этикет – это советы по уважительному общению с другими людьми. Именно он рассказывает, как правильно общаться со старшими, коллегами, как отвечать на неловкие вопросы. Все правила сводятся к формулам речевого этикета.
Правила общения касаются встречи (знакомства), общения при разговоре и его завершении. Они применимы к устной и письменной речи, официальным и речевым обращениям.
Функции речевого этикета
Речевой этикет делает общение приятным. Он нужен для вежливой беседы, правильным обращениям к старшим и руководящим должностям. Функции речевого этикета зависят от формы общения:
Слова и выражения для приветствий и прощаний, слова вежливости;
Обогащение речи, исключение слов-паразитов, мычаний и пауз;
Решение конфликтов спокойной и вежливой беседой;
Умение слушать собеседника до конца, не прерывая его вопросами и своими доводами.
История
Речевой этикет появился давно, когда люди только собирались в племена. Уже тогда к главам поселений и лекарям применялись вежливые формы обращения. К вождям, лекарям, воинам, жрецам были свои обращения, которые сохранились и сегодня.
Первым речевым этикетом были приветствия. Племена танцевали перед другими племенами, наклонялись или делали другие жесты. В Китае и Японии наклонялись со сжатыми ладонями, на Руси делали наклоны, и чем глубже, тем больше уважения было в жесте. Сейчас люди по всему миру жмут руки, целуют друг другу щеки, обнимают и похлопывают по спине.
Особенно популярны правила речевого поведения были у знати в XVII-XIX веках. После Октябрьской Революции универсальным вежливым обращением стали «товарищ» и «гражданин». До революции употребляли слова барин, барышня, государь. За границей популярны были слова сэр, милорд. Сейчас в уважительной форме принято говорить мисс, миссис, мистер, доктор и пр.
Сейчас в России и странах СНГ нет особых обращений. К незнакомым людям принято обращаться на «вы», «молодой человек», «девушка», «женщина», «мужчина».
Правила
Соблюдать правила речевого этикета просто и нужно, красивая и правильная речь вызывает симпатию у собеседника.
Вот самые простые правила речевого этикета:
Здороваться в полной форме: не «здрасьте», а «здравствуйте», употреблять слова добрый день и добрый вечер. С друзьями можно здороваться как угодно, но «привет» самый правильный вариант;
К незнакомым людям обращаться на «вы». На «ты» можно обращаться к другу, родственнику или к человеку, который сам вас об этом попросил. В официальной обстановке на «вы» нужно общаться со всеми;
Не называть человека по фамилии. Ровесника по имени, старшего по имени отчеству;
При завершении разговора прощаться, используя слова: до свидания, пока, до встречи. Будет уместно сказать, что общение понравилось, что было приятно провести время с человеком;
Не перебивать. Если появились вопросы, дослушайте собеседника до конца, быть может, он ответит на вопрос. Если нет, то задавать после паузы. Не перебивайте собеседника, чтобы рассказать похожий случай, приключившейся с вами. Если человек долго говорит, а у вас нет времени дослушать или вы чувствуете, что собеседник может продолжать еще долго, вежливо остановите его сказав, что послушали бы еще, но вам нужно бежать. Извинитесь за то, что перебили. Если собеседник потерял нить разговора, можете сказать, что он уклонился от темы;
Если нужно задать вопрос незнакомому человеку, говорите «извините пожалуйста» или «вы не могли бы сказать…». При любом ответе поблагодарите человека;
Первым протягивать руку для рукопожатия должен старший или человек с высшей должности.
Скачать доклад «Речевой этикет»
Скачать PDFРаспечатать
spadilo.ru
147 правил этикета, которые сделают вас лучше
Современный этикет — это правила поведения, помогающие производить приятное впечатление и строить эффективное общение. Они включают в себя опрятность, культуру речи, вежливость и умение держаться в различных ситуациях (за столом, в транспорте, в театре).
Современные правила этикета, которые сделают вас лучше
Азбука вежливости
При знакомстве представляют: мужчину — женщине, младших по возрасту и положению — старшим, пришедших позже — уже присутствующим. При этом человек, которому вы представляете незнакомца, упоминается первым, а тот, кого вы представляете, — вторым. «Анна, познакомься — это Константин», «Сергей Иванович, это Марина».
Знакомя людей, уместно давать краткие «справки» о них: «Это мой друг Никита, он хирург», «Это моя институтская приятельница Наталья». Так вы дадите понять, в каких отношениях состоите с каждым из собеседников, и предоставите им тему для беседы в ваше отсутствие: «…Так, значит, вы врач?».
Первыми приветствуют: мужчины — женщин, младшие по возрасту и положению — старших.
Но кто бы вы ни были — директор, академик, пожилая женщина или школьник, — входя в помещение, здоровайтесь первым.
Когда встречаются две супружеские пары, сначала здороваются друг с другом женщины, после этого мужчины приветствуют дам, затем здороваются между собой мужчины.
Руку для рукопожатия первым подаёт человек, которому представили незнакомца, то есть женщина — мужчине, старший — младшему и так далее. По правилам делового этикета первым всегда подаёт руку руководитель. Даже если подчинённый — женщина.
Если вам подали руку для рукопожатия, а вы сидите, встаньте.
Перед рукопожатием мужчина снимает перчатку. Женщине это делать необязательно.
Во время рукопожатия не курят. Если нет возможности выбросить сигарету, извинитесь за это.
Если руки чем-то заняты, рукопожатие неуместно. Поприветствуйте человека с тяжёлыми сумками или ребёнком на руках улыбкой и кивком головы во избежание неловкости и суеты.
В туалетной комнате руки не пожимают.
Руку в знак приветствия целуют лишь замужним женщинам и только в помещении. На деловых встречах и официальных мероприятиях целовать руки женщинам не принято.
Целуя руку женщине, наклоняйтесь, а не тяните руку к себе вверх.
Если вы идёте с кем-нибудь и ваш спутник поздоровался с незнакомым вам человеком, поздороваться следует и вам.
По улице мужчина должен идти слева от дамы. Справа могут идти только военнослужащие, которые должны быть готовы выполнить воинское приветствие.
Спускаясь по лестнице, мужчина идёт на одну-две ступени впереди женщины, а поднимаясь, — на одну-две ступени позади.
Кто бы вы ни были, всегда придерживайте дверь для идущих следом за вами.
Никогда не оборачивайтесь на оклик «Эй, ты!».
Никогда не показывайте пальцем на человека.
В разговоре с малознакомым человеком не стоит касаться личных тем: «Сколько вы зарабатываете?», «Слышал, ваша жена больна?», «Верите ли вы в Бога?». Удачные темы для непринуждённой беседы: спорт, погода, кулинария, домашние животные, искусство, наука, путешествия и тому подобное.
Стремясь блеснуть эрудицией, не переусердствуйте. Фразы вроде «Я это давно знаю», «Нашли чем удивить» и тому подобные лишь оттолкнут собеседника.
Правила поведения в театре, на концерте и в других общественных местах
Громко смеяться, шумно общаться, пристально разглядывать и обсуждать людей в публичном месте — оскорбительно.
Всегда включайте беззвучный режим или вовсе отключайте телефон в театре, библиотеке, кино, на лекции и так далее.
В кулуарах пользоваться телефоном позволительно. Но если вам нужно сделать или принять звонок, отойдите на два-три метра в сторону, чтобы не мешать беседе ваших друзей.
Курить в присутствии некурящих следует только с их разрешения.
Мужчина никогда не носит женскую сумку.
Женщина может не снимать в помещении шляпу и перчатки, но не шапку и варежки.
У зеркала в гардеробе можно осмотреть внешний вид и слегка поправить причёску. Но расчёсываться, красить губы, подтягивать брюки или чулки позволительно только в туалете.
Идите к своим местам в зале лицом к уже сидящим. Мужчина идёт первым.
Если в зрительном зале усаживаются две пары, женщины сидят в центре, мужчины — по обеим сторонам. В ложе впереди сидят женщины, за ними — мужчины.
В зрительном зале не стоит сидеть со склонёнными друг к другу головами. Даже если вы влюблены.
Оставьте поцелуи, объятия и прочие нежности на потом. Не заставляйте сидящих рядом людей лицезреть сразу два спектакля — на сцене и в зале.
На выставке не стремитесь прикоснуться к экспонатам руками, если это специально не разрешено.
Не пытайтесь сфотографироваться тайком там, где это запрещено правилами.
Правила поведения в транспорте
Садясь в такси, приветствуйте водителя первым. Правило не касается коллективного транспорта (автобуса, трамвая и прочего).
Место уступают: мужчины — женщинам, младшие по возрасту — старшим. Женщина в транспорте не уступает место мужчине, даже если он намного старше неё.
Никогда не занимайте специализированные места в общественном транспорте, если не имеете инвалидности и не беременны.
Следите за положением сидения и ног. Откинув спинку и вытянув ноги вперёд, вы будете ехать с комфортом. Но не ваши соседи.
Старайтесь в транспорте не разговаривать по мобильному телефону. Вряд ли вашим попутчикам интересны подробности вашей личной жизни. Возможно, они хотят вздремнуть или читают интересную книгу. Если кому-то нужно что-то срочно сообщить, напишите SMS.
Во время перелёта или поездки с детьми присматривайте за ними. Не позволяйте им шуметь, бегать по салону (вагону) и приставать к окружающим.
Если вы заметили в вагоне метро или салоне автобуса своего приятеля в час пик, просто поприветствуйте его кивком головы и улыбкой. Не стоит пробираться через толпу, причиняя неудобства другим пассажирам, чтобы «ехать вместе».
Мужчина выходит из общественного транспорта первым, чтобы помочь своей спутнице выйти и вынести багаж.
Правила поведения в гостях и на торжествах
Никогда не приходите в гости без звонка. А если вас навестили без предупреждения, можете позволить себе быть в халате и бигуди.
Отвечать на приглашение в гости вопросом «А кто ещё будет?» невежливо.
Принимая гостей, не оправдывайтесь: «У меня тут небольшой беспорядок, дети вечно всё разбрасывают», «У нас ремонт, поэтому везде коробки». Впуская человека в свой дом, вы выказываете ему своё доверие. Ваш гость должен уважать это и принимать ваше жилище таким, какое есть.
С приходом гостей следует выключить телевизор и отойти от компьютера.
Принимая гостей или находясь в гостях, некрасиво говорить по телефону больше 2–5 минут.
Преподнося хозяйке букет цветов, используйте правило: нечётно до дюжины. Если цветков больше 12, счёт идёт на полдюжины — число должно делиться на шесть. 13 цветков не дарят.
Не следует дарить юным девушкам цветы тёмных оттенков. Идеальный вариант в этом случае — белоснежный букет с нераскрывшимися бутонами. Женщинам в возрасте лучше не дарить быстро увядающие цветы. Зачем напоминать, что молодость уходит?
Если кто-то пришёл к вам впервые, сначала покажите гостю, где можно вымыть руки и привести себя в порядок, а затем пригласите в гостиную.
Когда начинать трапезу? Если за столом меньше восьми человек, ориентируйтесь на хозяина (хозяйку). Когда он сел и взял салфетку, можно приступать к еде.
Хозяйка не должна всё время бегать на кухню — об угощении следует позаботиться заранее. Мытьё посуды тоже подождёт.
На званый ужин или обед опаздывать нельзя. Если это случилось, нужно позвонить и предупредить хозяев, что задерживаетесь. На фуршет позволительно прийти позже назначенного часа и уйти раньше окончания мероприятия.
Если на фуршете подают шампанское или другие холодные напитки, то в начале мероприятия, когда все приветствуют друг друга, следует держать бокал в левой руке. От бокала ладонь может стать холодной и влажной, что нежелательно при рукопожатии.
Если во время фуршета вы беседуете с дамой, а официанты разносят аперитив, сначала поинтересуйтесь, что она предпочитает, а затем уже выберите напиток для себя.
Фуршеты или коктейльные вечеринки проводят в первую очередь для общения. Некрасиво всё время стоять возле стола с закусками и есть.
На торжественных приёмах мужчины, как правило, выслушивают тосты стоя. Женщинам вставать необязательно.
Если вы уходите с приёма раньше остальных гостей, прощайтесь только с хозяевами встречи. Иначе ваш уход может послужить для участников вечеринки сигналом к тому, что всем пора по домам.
Правила этикета за столом, в ресторане
Фраза «Я вас приглашаю» означает, что вы платите. Если звучит «Давайте сходим в ресторан», каждый платит за себя. Но даже в этом случае мужчина может предложить спутнице расплатиться за неё.
Условившись о встрече с женщиной в кафе или ресторане, мужчина обязан быть на месте несколько раньше, чтобы успеть найти свободный столик.
Если в ресторане предусмотрено только одно меню на столик, мужчина предлагает его даме, которая в свою очередь может попросить спутника прочесть меню вслух. В большой компании меню для всех читает кто-нибудь один.
Подсесть к знакомому, сидящему в кафе в обществе незнакомого вам человека, допустимо. Но не в ресторане. Исключение — если сидящие просят вас об этом.
Нельзя просить счёт, когда партнёрша или партнёр ещё едят.
За столом категорически нельзя: скатывать хлебные шарики, играть приборами и посудой, сворачивать край скатерти в трубочку, протягивать ноги под столом во всю длину.
За столом следует опираться на его край только запястьями. Женщинам позволительно ненадолго поставить на стол локти.
Салфетку складывают вдвое сгибом к себе и кладут на колени, а не закладывают её за воротник.
Недопустимо использовать салфетку вместо носового платка.
Перед тем как попить, желательно промокнуть рот уголком салфетки, чтобы не оставлять на бокале жирных следов. Женщина с накрашенными губами должна вытирать рот бумажной, а не полотняной салфеткой.
Если необходимо выйти из-за стола, салфетку кладут на стол слева. Но в Европе, например, её оставляют на спинке стула. По окончании обеда салфетку кладут справа от тарелки.
Если салфетка упала на пол, не следует поднимать её самостоятельно. Это сделает официант, он же принесёт вам новую.
Чтобы случайно не покуситься на хлеб или вино соседа, тарелка для хлеба располагается слева, напитки — справа. Это легко запомнить: сложите руки в два жеста OK, при этом слева получится буква b (bread — хлеб), справа — d (drink — напиток).
Положение приборов на тарелке может служить сигналом для официанта: «Закончил трапезу, можно убирать», «Ещё ем», «Жду следующее блюдо» и так далее.
Ножом следует лишь резать пищу, а не класть её в рот.
Если к блюду подают соус в отдельной посуде, его следует понемногу класть на свою тарелку. Ни в коем случае не макайте куски в общую соусницу.
Не следует промакивать соус в тарелке хлебом. Но, если вы не в силах удержаться, помогайте себе вилкой, а не пальцами.
Невежливо выбирать из порций, лежащих на блюде, лучшую. Берите кусок, который ближе к вам.
Некрасиво доливать из бутылки или графина только себе. Взяв сосуд в руки, сначала предложите напиток соседу по столу.
Если вы пьёте напиток через соломинку, не высасывайте всё до последней капли. Пронзительное бульканье на дне стакана — не самый приятный звук для окружающих.
Блюдце не берут в руки, поднимают только чашку.
Никогда не используйте ноготь мизинца в качестве зубочистки.
Правила этикета в интернете
Не пренебрегайте темой при отправлении email. Она должна отражать суть письма и тем самым экономить время собеседника.
Подтверждайте получение важных писем, если ответ на них требует времени.
Использовать тяжёлые вложения в письмах — всё равно что путешествовать с громоздким багажом. Неудобно всем: и адресату, и вам самому.
Общение в мессенджере может быть синхронным и асинхронным. Чаще всего обмен сообщениями происходит мгновенно. Но не стоит обижаться, если сообщения приходят «с перебоями». Вероятно, человек просто занят.
Использование CapsLock приравнивается к крику.
Не злоупотребляйте кнопкой «Отправить». Дробя длинное сообщение на куски, вы усложняете взаимопонимание. Человек может решить, что мысль закончена и начать отвечать. Причём, если он будет это делать так же, как вы, постоянно нажимая на Enter, вы запутаетесь.
Если во время беседы собеседник вдруг стал offline, это не повод для обиды.
Если «стучитесь» к кому-то в друзья в социальной сети, напишите, кто вы и почему хотите добавиться.
Не постите на стене друга слишком личные сообщения и фотографии. Социальные сети — не место для проявления интимных чувств и выяснения отношений. Лучше делать это лично.
Спрашивайте разрешение на публикацию фотографии другого человека в социальной сети. Даже если это ваш закадычный друг.
Не злоупотребляйте #хэштегами.
Возьмите под контроль приглашения и уведомления, рассылаемые от вашего имени в социальных сетях. Не засыпайте друзей спамом.
Если вы получили сообщение, но в силу каких-то причин не могли на него ответить сразу (например, были в оффлайне) – желательно ответить в течение суток. Даже если вы видите, что собеседник не в сети – все равно не затягивайте с ответом.
Если в вашей программе для диалогов горит статус «Онлайн», «Активен», «Доступен» — отвечать следует в течение 5 минут после получения запроса. В случае, если вы ведете длинный диалог с клиентом по телефону или отошли на обед – установите статус «Недоступен», «Занят», «Отошел». Как только появится возможность – сразу же ответьте на входящие сообщения.
Помните о том, что собеседник находится в офисе – он может говорить по телефону, общаться с руководством компании или клиентами. Имейте терпение, не «теребите» партнера по бизнесу или коллегу каждую минуту – дождитесь, пока он сможет ответить. В том случае, если у собеседника горит статус «Занят», но у вас есть особо срочный вопрос – поинтересуйтесь, когда человек сможет уделить вам внимание. Это вполне соответствует правилам этикета.
Если ответ собеседника вам важен именно сегодня – старайтесь писать ему в удобное (рабочее) время, чтобы человек успел подготовить нужную информацию.
Если у вас серьезная тема к обсуждению – не пишите ее в виде набора коротких сообщений, это мешает точно понять суть вопроса. Даже если вы хотите как можно быстрее объяснить ситуацию – напишите и отправьте подробное сообщение. Это займет у вас чуть больше времени, но зато партнеру проще воспринять информацию.
Не требуйте ответ мгновенно – дайте собеседнику время на размышления. Даже если у человека статус «Онлайн» — это не значит, что партнеру не нужно «переварить» информацию, выбрать наиболее верный вариант ответа. Не переспрашивайте, не забрасывайте собеседника другими вопросами – и тогда общение будет легким, понятным обеим сторонам.
После написания письма перечитайте его и проверьте орфографию.
Будьте терпимы к другим людям, и если ваш собеседник не соблюдает правила этикета, не стоит общаться в его манере.
Старайтесь также не пользоваться сокращениями «имхо», «спс», «пжлст», «пжт». Это очень некультурная и неуважительная форма общения.
Не жалуйтесь постоянно на что-либо. Не стоит писать только о негативе, иногда и вам, и собеседнику будет гораздо приятнее обсудить что-то положительное.
Не рассказывайте слишком много о себе. Часто в социальных сетях можно прочитать практически всю информацию о жизни пользователя, причем дополненную фотографиями, это не только не очень красиво, но и опасно, так как посторонним людям будет доступна вся информация о вас.
Не будьте пафосными. Не делайте из своей стенки в социальной сети доску для публикации пафосных высказываний о политике и жизни. Если вы ведете группу, то не относитесь к читателям свысока.
Не используйте малоизвестную лексику. Конечно, хорошо, что вы знаете много терминов и определений, но не стоит их употреблять в диалоге с теми людьми, кто с ними не знаком.
Общение в сети мало чем отличается от реального. По сути, эти правила этикета для интернета ничем не отличаются от общепринятых норм поведения, только вносят некоторые дополнения, обусловленные особенностями виртуального общения: со старшими нужно общаться на «вы», так же как и с незнакомцами.
Правила этикета в лифте
При ожидании лифта мужчиной и женщиной, первым входит в лифт мужчина. Если лифт ожидает группа людей, то первыми заходят те, кто ближе стоит к двери. В домах и учреждениях, где люди знают друг друга, вперед пропускают того, кто едет дальше.
Ребенок всегда заходит в лифт после взрослого, а выходит перед ним.
Мужчина, сопровождающий даму, нажимает кнопку нужного этажа. Так же он поступает, если в лифте кроме него едут женщины и люди старшего возраста. В переполненной кабине на кнопки нажимает тот, кто стоит рядом, остальные просят его об этом, называя этажи.
По правилам хорошего тона женщине не стоит тянуться к кнопкам, если рядом с ними стоит мужчина, даже посторонний. Предпочтительнее попросить его нажать кнопку нужного этажа.
В тесном замкнутом пространстве лифта надо соблюдать особую деликатность. Некрасиво смотреть на других пассажиров в упор или разглядывать их украдкой. Лучше всего смотреть вниз, либо обратиться к стене. Однако, обмениваясь репликами, пассажирам лифта следует смотреть друг другу в глаза.
При любом прикосновении нужно немедленно извиниться независимо от того, кто был его виновником. Если вам отдавили ногу, примите извинения и скажите: «Пожалуйста».
Пропуская выходящих, лучше выйти из лифта, чем прижиматься к стенке и создавать толкучку.
В лифте здороваются со знакомыми, но разговаривать без острой необходимости не принято, поскольку другие пассажиры становятся невольными слушателями вашего диалога.
Этикет разрешает мужчине не снимать головной убор в лифте. И все же настоящий джентльмен, сопровождая дам, непременно снимет шляпу, если обстоятельства позволяют это сделать.
Правила офисного этикета
Мойте за собой посуду после обеда. Сразу мойте. Лучше вообще выкинуть ланчбокс, чем оставлять его грязным в раковине.
Не снимайте обувь. Переобувайтесь в местах, которые для этого предназначены. Ну или хотя бы достаточно удалены от чужих столов. И ноги на стол можно класть, только если вы уходите последним, а рядом уже никого нет.
Прежде чем бежать в IT-отдел, перезагрузите компьютер.
Не трогайте чужой йогурт. И чужое яблоко. Чтобы взять печенье, тоже нужно разрешение.
По утрам начальству нужно говорить «Здравствуйте», а не «Здорово, чувак!», если, конечно, начальник сам так не говорит.
Если вы с коллегой оказались в соседних кабинках в туалете и вы закончили раньше, не надо дожидаться, когда он закончит, чтобы вместе вернуться в офис.
Когда в коробке с канцтоварами осталось всего три пачки стикеров, не забирайте себе всё, лучше положите две на столы коллег. Немного заботы в офисе.
Рабочий стол должен выглядеть как аккуратный подстриженный газон, а не как непролазная чаща.
Подошли к кулеру, наполнили стакан, отошли от кулера. Не надо стоять перед ним и общаться с коллегами. Если поговорить очень хочется, хотя бы сделайте пару шагов в сторону и освободите для других дорогу к воде.
За столом не рыгать, если только все коллеги не сидят в наушниках.
Ланчбоксы и одноразовые коробки с едой подписывайте. Не обязательно своим именем, можно и ник использовать. Например, «Царь, просто царь». Такое точно не выкинут.
Здоровайтесь со всеми, даже если вы работаете в разных концах офиса и понятия не имеете, как этих сослуживцев зовут.
На совещаниях отводится время для вопросов. Вот тогда их и задавайте, не перебивайте докладчиков.
Не надо делать украшения из скрепок.
Приходя на работу, переключайте телефон на виброзвонок и не оставляйте устройство включённым на столе (в сумке).
Рукопожатие — вещь необязательная, но принятая в деловом сообществе.
Заканчивает переписку тот, кто её начал, т. е. последнее письмо должно исходить от того, кто написал первым.
В телефонных разговорах действует правило — если вы звоните начальнику, то первым трубку кладёт именно он.
Не надо есть в офисе еду с резкими запахами, при всей своей любви к селёдке, квашеной капусте, чесноку и котлеткам старайтесь обходиться на работе без них.
Если на рабочем месте вы пьёте только чай и кофе, не ставьте чашку на документы, поскольку на бумаге может остаться след, который явно будет говорить не в вашу пользу.
Вы, безусловно, имеете право пить различные напитки в течение дня, но кружка при этом должна выглядеть опрятно — никаких многократно заваренных пакетиков чая, следов от губной помады с внешней стороны и подобных вещей быть не должно.
Воспитанные люди не проходят в верхней одежде на своё рабочее место, не вешают её на спинку стула и, тем более, не кладут на стол. Для этого есть гардероб. Единственное исключение — когда вы забежали в офис буквально на 5–10 минут и потом опять куда-то уйдёте. Такой вариант позволителен.
Хорошие манеры и технологии
Люди важнее технологий! Не кладите смартфон на стол в общественных местах. Поступая так, вы показываете, насколько важную роль играет в вашей жизни телефон и насколько неинтересно вам происходящее вокруг. В крайнем случае, если ждёте важного звонка, достаньте телефон и положите на стол экраном вниз.
На звонок лучше отвечать после второго-третьего звонка, чтобы звонящий успел сосредоточиться. Если вам не ответили после пятого гудка, положите трубку.
Если во время звонка прервалась связь, перезвонить должен инициатор разговора.
Называйте своё полное имя, если звоните с незнакомого номера.
Сообщать о важных событиях по SMS неприлично.
Предупреждайте о видеозвонке текстовым сообщением. Не ставьте ни собеседника, ни себя в неловкое положение.
Во время видеозвонка общайтесь с человеком, а не любуйтесь собственным изображением в углу экрана. Камера — это не зеркало в ванной.
Всегда соблюдайте тайну переписки. Не читайте чужих писем, не заглядывайте в монитор, когда кто-то общается в чате, отворачивайтесь, когда человек набирает пароль.
Обращайтесь с людьми так, как хотите, чтобы обращались с вами!
Прочитали? А теперь проверьте свои знания в одном из тестов: «Тест на знание этикета», «Как хорошо вы знаете базовые правила этикета?», «Знакомы ли вы с правилами хорошего тона?».
Задачи про рыцарей и лжецов в 4-5 классе. Задачи с решениями онлайн
Рассказываем, как в 4-5 классе решать задачи про рыцарей и лжецов, в которых надо определить, кто является лжецом, а кто говорит правду.
Схема решения этих задача такова — разобрать все случаи и отсеять те, что приводят к противоречию. Рыцари всегда говорят правду, а лжецы лгут.
Задача 1
В одном городе живут рыцари и лжецы. Рыцари всегда говорят правду, а лжецы всегда обманывают. Путешественник встретил двух жителей этого города. Один из них сказал: «По крайней мере один из нас лжец!». Кто этот горожанин — рыцарь или лжец? Кто второй горожанин?
Решение
Если разговорчивый горожанин лжец, то фраза «По крайней мере один из нас лжец!» является неправдой, то есть среди них нет ни одного лжеца. А это приводит к противоречию, т.к. он лжец. Если же он сказал правду, то второй горожанин — лжец.
Ответ: первый — рыцарь, второй — лжец.
Задача 2
В одном городе живут рыцари и лжецы. Рыцари всегда говорят правду, а лжецы всегда обманывают. Горожанин Пит сказал своим друзьям: — Вчера мой сосед заявил мне, что он лжец! Кем является Пит — рыцарем или лжецом?
Решение
1 способ
Лжец не может заявить, что он лжец, так как это будет правдой, а это противоречие, следовательно, Пит лжет.
2 способ
Можно рассмотреть два случая.
1. Если сосед Пита — рыцарь, тогда то, что он заявил Питу, должно быть правдой, то есть он должен быть лжецом. Но мы предположили, что он рыцарь. Значит, такого не может быть.
2. Если сосед Пита — лжец, то он сказал Питу неправду, то есть неправда, что он лжец. Снова противоречие.
Итак, если бы сосед Пита сказал ему, что он лжец, то в любом случае получилось бы противоречие, то есть, такого быть не могло. Вывод: сосед Пита этого вообще не говорил!
Значит, Пит лжёт.
Ответ: Пит — лжец
Хотите, чтобы ваш ребёнок обучался самостоятельно? Вам поможет наш ВИДЕОКУРС
Задача 3
Есть два поселка, которые разделяет мост. С одной стороны моста в поселке Правдорубово живут рыцари, которые говорят только правду, с другой в поселке Честновруново лжецы, которые всегда врут. Жители двух поселков любят ходить друг к другу в гости. Путешественник оказался в одном из этих поселков, он задал вопрос первому встретившемуся человеку: «Это ваш родной поселок?». На что получил ответ: «Нет, я здесь в гостях». В каком поселке оказался путешественник?
Решение
Первый встречный мог быть либо рыцарем, либо лжецом. Если он рыцарь, то его ответ правдив, и он не живёт в этом поселке, и тогда это поселок лжецов. Если же он лжец, то его ответ — ложь, и он живет в этом поселке а значит, это опять же поселок лжецов. Итак, в обоих случаях получается, что путешественник оказалась в поселке лжецов Честновруново.
Ответ: в поселке лжецов Честновруново.
Задача 4
Встретились два горожанина Тыс и Мыс. — По крайней мере один из нас – рыцарь, — глубокомысленно изрек Тыс. — Но ты то уж точно лжец! – рассмеялся ему в лицо Мыс. Определите, кем являются оба?
Решение
Если предположить, что Мыс рыцарь, то он говорит правду и Тыс лжец, но этого не может быть, т.к. Тыс в таком случае сказал правду, что один из них рыцарь. Следовательно, Мыс лжет, а Тыс рыцарь. Это подтверждается их высказываниями.
Ответ: Тыс – рыцарь, Мыс – лжец
Задача 5
В одном городе живут рыцари и лжецы. Рыцари всегда говорят правду, а лжецы всегда обманывают. Горожанин Марк выкрикнул: « Или я лжец, или Артур рыцарь!». Кто такой Марк (рыцарь или лжец), и кто такой Артур?
Решение
В высказывании Марка стоит противопоставление: либо одна либо другая часть правда. Я — лжец, лжец про себя так не скажет, т.к. он будет всегда лгать, следовательно, Марк — рыцарь и первая часть его высказывания неверна, а вторая верна, т.е. Артур тоже рыцарь.
Ответ: Марк и Артур оба рыцари.
Задача 6
В одном городе живут рыцари и лжецы. Рыцари всегда говорят правду, а лжецы всегда обманывают. Горожанин Бук высказал утверждение: «Я лжец, а Тук не лжец». Кто такой Бук (рыцарь или лжец), и кто такой Тук?
Решение
Здесь во фразе нет противопоставления, выражение должно быть либо правдой, либо ложью. Рыцарь про себя не может сказать, что он лжец, следовательно, Бук – это лжец. Так как он сказал про себя правду в первой части фразы, то во второй он должен наврать, чтобы высказывание стало ложью, следовательно, Тук тоже лжец.
Ответ: Бук и Тук оба лжецы.
Задача 7
Однажды в четверг после дождя между горожанами Тимом и Бимом произошел следующий диалог: — Ты можешь сказать, что я рыцарь, — гордо заявил Тим. — Ты можешь сказать, что я лжец, — грустно ответил ему Бим. Кем являются Тим и Бим?
Решение
Пусть Тим — рыцарь, то есть говорит правду. Тогда Бим может сказать, что Тим рыцарь. Поскольку это правда, то получается, что Бим может сказать правду, значит, Бим тоже рыцарь. Но тогда сказанное Томом тоже должно быть правдой, но на самом деле Тим не сможет сказать, что он лжец, потому что он не лжец, а Тим не врёт. Противоречие.
Значит, Тим — лжец. Тогда Бим не может сказать, что он рыцарь, то есть Бим не может сказать неправду. Значит, Бим рыцарь. И действительно, слова Бима — правда, потому что Тим может соврать, сказав, будто Бим лжец.
Ответ: Тим – лжец, Бим – рыцарь.
ВИДЕОКУРС 2plus2.online по решению олимпиадных задач по математике для 4 класса и задач из вступительных экзаменов в 5-й класс физматшколы.
Задача 8
Волшебная страна населена лжецами и рыцарями, причем лжецы всегда лгут, а рыцари всегда говорят правду. Путешественник едет по этой стране в сопровождении местного гида. Навстречу им попадается туземец и путешественник спрашивает его: «Вы, конечно, рыцарь?» Туземец его понимает и отвечает «Гашака», что означает то ли «да», то ли «нет». На просьбу перевести гид говорит: «Он сказал — да. Добавлю, что на самом деле он лжец». Кем, на самом деле, был встреченный ими туземец?
Решение
Заметим сначала, что путешественник и без помощи гида мог определить, что слово «Гашака» обозначает «да». Действительно, независимо от того, является ли житель страны рыцарем или лжецом, на вопрос «Вы, конечно, рыцарь?» он ответит «да». Значит, гид является рыцарем и говорит правду, но из его слов следует, что туземец — лжец. Следовательно, так оно и есть.
Ответ: Лжецом.
Задача 9
В городе «Правдивая ложь» живут рыцари и лжецы. Рыцари всегда говорят правду, лжецы всегда лгут. Странствующий путник встретил троих горожан и спросил каждого из них: «Сколько рыцарей среди твоих друзей?». Первый ответил: «Ни одного». Второй сказал: «Один». Что сказал третий горожанин?
Решение
Если первый — рыцарь, то, по его словам, второй и третий — лжецы. Но это невозможно из-за высказывания второго горожанина. Так получилось бы, что второй сказал бы правду, хотя он лжец. Значит, первый — лжец. Если второй — лжец, то по его словам третий тоже лжец. Но тогда первый горожанин сказал правду, а он должен был соврать. Значит, второй — рыцарь. И, по его словам, третий тоже рыцарь. Третий честно ответит: «Один».
Ответ: Один.
ВИДЕОКУРС 2plus2.online по решению олимпиадных задач по математике для 4 класса и задач из вступительных экзаменов в 5-й класс физматшколы.
Задача 10
Встретились трое горожан из города, в котором живут рыцари и лжецы. Горожанин Ван говорит: «Мы все трое лжецы». Ман говорит: «Ровно один из нас троих рыцарь». Тан молчит. Кто из них кто?
Решение
Ван не может быть рыцарем, ибо тогда они все (включая и его) были бы лжецами. Поэтому Ван – лжец. Поэтому он лжет, и среди них троих есть рыцари (хотя бы один). Если Ман – рыцарь, то он говорит правду, и Тан тогда лжец. Если же Ман – лжец, то рыцарей среди них либо нет, либо 2 или 3. Последние два варианта, очевидно, невозможны (Ман и Ван уже лжецы). Поэтому тогда и Тан должен быть лжецом. Но это противоречит ранее доказанному, что среди них есть рыцари. Поэтому Ман не может быть лжецом.
Ответ: Ван – лжец, Ман – рыцарь, Тан– лжец.
Задача 11
Путешественник встретил двух жителей города, в котором живут лжецы и рыцари. Он спросил обоих: «Рыцарь ли его приятель?» и получил ответы. Должны ли оба ответа быть одинаковыми?
Решение
Если оба встретившихся вам островитянина рыцари, то они оба ответят «да». Если они оба лжецы, то они также оба ответят «да». Если же один из них рыцарь, а другой лжец, то рыцарь ответит «нет» и лжец также ответит «нет».
Ответ: Должны.
Задача 12
Встретились два горожанина Том и Сем. Том заявил: «Мы оба лжецы, и этот город называется Троя». Сем ответил: «По крайней мере один из нас лжец, и этот город не Троя». Возможно ли, чтобы этот город действительно назывался Троя? Если да, то наверняка ли он так называется?
Решение
Пусть этот город действительно называется Троя. Тогда вторая часть высказывания Тома истинна, вторая часть высказывания Сема – ложна. Том не может быть рыцарем, так как рыцарь не скажет о себе, что он лжец. Значит, Том – лжец. То есть, первая часть высказывания Сема истинна, а, так как вторая ложна, то ложно и все его высказывание в целом. То есть Сем – лжец. Значит, они оба лжецы. Но тогда обе части высказывания Тома истинны, что невозможно, следовательно, Том – лжец). Значит, этот город не может называться Троя. Получается, что Том лжец, а Сем – рыцарь.
Ответ: нет, нельзя.
Дата публикации
Купить наш видеокурс по подготовке к поступлению в 5-й класс физматшкол и участию в математических олимпиадах
Как готовиться к математическим олимпиадам и поступлению в физматшколу
Задачи раздела:
Более лёгкие задачи находятся внизу списка.
В парламенте города М — 101 депутат. Среди них есть правдорубы и лжецы
В парламенте города М — 101 депутат. Среди них есть правдорубы, которые всегда говорят правду и лжецы, которые всегда лгут. В условиях финансового кризиса было принято решение сократить парламент на одного депутата. Но каждый из депутатов заявил, что если его выведут из состава парламента, то среди оставшихся депутатов большинство составят лжецы. Сколько правдорубов и сколько лжецов в парламенте?
Подробно
Однажды Алиса оказалась в волшебной стране, где есть город рыцарей и город лжецов
Однажды Алиса оказалась в волшебной стране. В этой стране есть всего два города: город рыцарей и город лжецов. Жители города лжецов всегда лгут, а жители города рыцарей всегда говорят правду. Притом все они часто ездят друг к другу в гости. Алиса спросила первого встречного, живёт ли он в этом городе? «Нет, я здесь в гостях», — ответил тот. В каком городе была Алиса?
Подробно
В стране лжецов и рыцарей десяти жителям выдали различные числа от 1 до 10
В стране лжецов и рыцарей (рыцари всегда говорят правду, лжецы всегда лгут) десяти жителям выдали различные числа от 1 до 10. Потом каждого спросили: «Делится ли ваше число на 2?». Утвердительный ответ дали 3 человека. На вопрос «Делится ли ваше число на 4?» утвердительный ответ дали 6 человек. На вопрос «Делится ли ваше число на 5?» утвердительно ответили 2 человека. Сколько было лжецов и какие у них были числа?
Подробно
В клетках квадрата 4×4 стоят горожане (рыцари и лжецы)
В клетках квадрата 4×4 стоят горожане (рыцари и лжецы). В некоторый момент каждый из них произнес: «Во всех соседних со мной клетках стоят лжецы». Какое наибольшее количество лжецов могло быть среди них?
Подробно
За круглым столом сидят 10 человек – рыцари и лжецы
За круглым столом сидят 10 человек – рыцари и лжецы (рыцари всегда говорят правду, а лжецы всегда лгут). Известно, что у каждого из них за этим же столом есть ровно один друг, причем у рыцаря этот друг – лжец, а у лжеца этот друг – рыцарь (дружба всегда взаимна). Каждого сидящего за столом спросили: «Сидит ли рядом с вами ваш друг?». Некоторые ответили: «Да». Сколько таких могло быть?
Подробно
Все жители города рыцерй и лжецов стали в круг лицом к центру, и каждый сказал путешественнику про соседа справа, рыцарь ли он
Путешественник посетил город, каждый житель которой либо рыцарь (всегда говорит правду), либо лжец (всегда лжёт). Всего в городе живёт 26 человек. Все жители города стали в круг лицом к центру, и каждый сказал путешественнику про соседа справа, рыцарь ли он. На основании этих сообщений путешественник смог однозначно определить количество лжецов и рыцарей в городе. Сколько лжецов живёт в этом городе?
Подробно
В городе живут 50 рыцарей и 50 лжецов, а больше никто не живёт
В городе живут 50 рыцарей и 50 лжецов, а больше никто не живёт. Рыцари говорят только правду, а лжецы всегда лгут. У каждого рыцаря есть хотя бы один друг, а у каждого лжеца ровно один друг. Как-то раз каждый произнес фразу «Ни один из моих друзей не является лжецом», либо «Ни один из моих друзей не является рыцарем», причем каждую из фраз произнесло ровно 50 человек. Какое наименьшее возможное число пар друзей, один из которых рыцарь, а другой — лжец?
Подробно
В городе живут 100 рыцарей и 100 лжецов, у каждого из них есть хотя бы один друг
В городе живут 100 рыцарей и 100 лжецов, у каждого из них есть хотя бы один друг. Рыцари всегда говорят правду, а лжецы всегда лгут. Однажды утром каждый житель произнес фразу «Все мои друзья — рыцари», либо «Все мои друзья — лжецы», причем каждую из фраз произнесло ровно 100 человек. Найдите наименьшее возможное число пар друзей, один из которых рыцарь, а другой — лжец.
Подробно
По кругу сидят рыцари и лжецы – всего 12 человек
По кругу сидят рыцари и лжецы – всего 12 человек. Каждый из них сделал заявление: «Все кроме, быть может, меня и моих соседей – лжецы». Сколько рыцарей сидит за столом, если известно, что лжецы всегда врут, а рыцари всегда говорят правду?
Подробно
В городе «Лживая правда» живут рыцари и лжецы, всего 32 жителя
В городе «Лживая правда» живут рыцари и лжецы, всего 32 жителя. Рыцари всегда говорят правду, а лжецы всегда лгут. Однажды все жители города собрались на центральной площади, и каждый житель сказал: “Все вы лжецы”. Сколько в городе лжецов?
Подробно
В городе, где живут рыцари и лжецы, встретились 13 жителей
В городе, где живут рыцари и лжецы, первые всегда говорят правду, а вторые вру, встретились 13 жителей. Каждый из них заявил остальным: «Вы все – лжецы!» Сколько рыцарей было среди этих тринадцати жителей?
Подробно
В маленьком городе под названием «Правдивая ложь» живут лжецы и рыцари, всего 21 человек
В маленьком городе под названием «Правдивая ложь» живут лжецы и рыцари, всего 21 человек. Рыцари всегда говорят правду, а лжецы всегда лгут. Каждый житель города заявил: «Среди оставшихся горожан более половины — лжецы». Сколько лжецов в городе?
Подробно
На затерянном острове, населенном рыцарями и лжецами, разнесся слух о том, что на нем зарыты сокровища
На затерянном острове, населенном рыцарями и лжецами, разнесся слух о том, что на нем зарыты сокровища. Вы прибываете на остров и спрашиваете у одного из местных жителей, есть ли золото на его острове. В ответ на ваш вопрос туземец заявляет: «Сокровища на этом острове есть в том и только в том случае, если я рыцарь». Есть ли сокровище на этом острове? Можно ли определить, кто этот туземец — рыцарь или лжец?
Подробно
Встретились трое горожан A, B и C, о каждом из которых известно, что он либо рыцарь, либо лжец, либо хитрец
В городе живут рыцари, которые говорят всегда правду, лжецы, которые всегда лгут и хитрецы, которые могут лгать и говорить правду. Встретились трое горожан A, B и C, о каждом из которых известно, что он либо рыцарь, либо лжец, либо хитрец. A говорит: «B по рангу выше, чем C.». B говорит: «C по рангу выше, чем A.». Затем у C спрашивают: «Кто старше по рангу — A или B?» Что ответит C?
Подробно
Про браки между рыцарями, лгунами и хитрецами
По древней традиции в стране, где живут рыцари, лгуны и хитрецы брак разрешен только между рыцарем и лгуньей или между двумя хитрецами. В одной супружеской паре мистер Хек высказал следующие утверждения: «Моя жена — не хитрец», а миссис Хек заявила: «Мой муж — не хитрец». Кто такой мистер Хек и кто такая миссис Хек — рыцарь, лжец или хитрец?
Подробно
Встретились трое горожан рыцарь, лгун и хитрец
В городе живут рыцари, лжецы и хитрецы. Рыцари всегда говорят правду. Лжецы всегда лгут. А хитрецы могут как соврать, так и сказать правду. Встретились трое горожан рыцарь, лгун и хитрец. Кто из них кто, если A заявил: «Я хитрец», на что B согласился: «Да, А хитрец», а С сказал: «Я не хитрец».
Подробно
На острове живут рыцари, лжецы и хитрецы
На острове живут рыцари, лжецы и хитрецы. Рыцари всегда говорят правду. Лжецы всегда лгут. А хитрецы могут как соврать, так и сказать правду. Жителя острова однажды спросили: “Вы лжец?”. А он ответил: “Да, я лжец.” Кто был этот человек?
Подробно
Проводник для похода через лес не должен оказаться оборотнем — 3
Вам нужно нанять проводника для похода по лесу в полнолунье среди местных жителей, и он не должен оказаться оборотнем. Все местные жители делятся на лжецов, которые всегда лгут, и рыцарей, которые всегда говорят правду. Оборотнем может быть, как рыцарь, так и лжец. Известно, что по крайней мере один из них оборотень и ни один не является одновременно рыцарем и оборотнем. A заявил: «По крайней мере один из нас рыцарь». B сказал: «По крайней мере один из нас лжец». С промолчал. Кто оборотень А, В или С?
Подробно
Проводник для похода через лес не должен оказаться оборотнем — 2
Вам нужно нанять проводника для похода по лесу в полнолунье среди местных жителей, и он не должен оказаться оборотнем. Все местные жители делятся на лжецов, которые всегда лгут, и рыцарей, которые всегда говорят правду. Оборотнем может быть, как рыцарь, так и лжец. Знакомый рыцарь вам шепнул, что один из претендентов точно оборотень. Первый претендент заявил вам: «Оборотень – рыцарь». Второй сказал: «Оборотень – лжец». Кого вы возьмете в проводники?
Подробно
Проводник для похода через лес не должен оказаться оборотнем — 1
Вам нужно нанять проводника для похода по лесу в полнолунье среди местных жителей, и он не должен оказаться оборотнем. Все местные жители делятся на лжецов, которые всегда лгут, и рыцарей, которые всегда говорят правду. Оборотнем может быть как рыцарь, так и лжец. Знакомый рыцарь вам шепнул, что один из претендентов точно оборотень. В беседе с вами они заявляют: A: «C – оборотень». B: «Я не оборотень». C: «По крайней мере двое из нас лжецы». Кто оборотень: рыцарь или лжец? И кого вы возьмете себе в проводники горожанина А, В или С?
Подробно
В городе лжецов и рыцарей завелись оборотни, они могут быть как рыцарями, так и лжецами
В городе лжецов и рыцарей завелись оборотни, они могут быть как рыцарями, так и лжецами, а в полнолунье они превращаются в волков и пожирают людей. Напомним, что рыцари всегда говорят правду, а лжецы всегда лгут. Под подозрение попали три горожанина: А, В, С. Среди них имеется ровно один оборотень. А говорит: «Я оборотень». В говорит: «Я оборотень». С говорит: «Не более чем один из нас рыцарь». Кто оборотень? Можно ли установить, кем являются А, В, С (рыцарями или лжецами)?
Подробно
Перед судом предстали три горожанина
Некогда перед судом предстали три горожанина, которых для конфиденциальности мы обозначим А, Б и В. Известно, что преступление совершил ровно один из них, но кто из них является рыцарем, а кто – лжецом, было неизвестно (одни всегда говорят правду, а другие лгут). А заявил, что Б лжец. Но преступление совершил В. Б заявил, что А и В либо оба рыцари, либо оба лжецы. В высказался, что Б говорит правду. Но тем не менее он и совершил преступление. Помогите судье определить кто есть кто (лжецы и рыцари), и кто совершил преступление.
Подробно
Путешественник спросил у двух горожан: «Кто-нибудь из вас рыцарь?»
В городе лжецов и рыцарей путешественнику встретились два горожанина. Он спросил у одного из них: «Кто-нибудь из вас рыцарь?» Его вопрос не остался без ответа, и он узнал то, что хотел узнать. Кем был горожанин, к которому он обратился с вопросом рыцарем или лжецом? Кем был другой горожанин?
Подробно
Каждый горожанин отвечал путешественнику: «Лжец выше меня ростом!»
В городе живут рыцари и лгуны. Рыцари всегда говорят правду, а лжецы лгут. Прибывший в город путешественник спросил: «Кто из вас лжец?» И каждый горожанин ему отвечал: «Лжец выше меня ростом!» Сколько живет в городе лжецов?
МБОУ «Заинская средняя общеобразовательная школа № 3»
Заинского муниципального района Республики Татарстан
Руководитель: Хасметдинова А. А.
учитель математики
МБОУ «Заинская средняя общеобразовательная школа № 3»
Заинского муниципального района Республики Татарстан
Заинск, 2019
Оглавление
Введение
Подготовка к исследованию
Исторические сведения
Проведение исследования
2.1 Рассмотрение логических задач
2.2 Исследование уровня логического мышления
2.3. Социальный опрос
Выводы
Заключение
Список литературы
Введение
Впервые с решением нестандартных задач мы столкнулись во время олимпиады по математике, нам стало интересно, а как бы справились с решением подобных задач мои одноклассники и ученики других классов. Поэтому мы решили провести собственное исследование данной темы и назвали ее «В стране рыцарей и лжецов». При решении логических задач предоставляется возможность подумать над необычным условием, рассуждать. Решать логические задачи очень увлекательно! Задачи на нестандартное логическое мышление помогут и в повседневной жизни решать проблемы нестандартным образом.
Актуальность. В наше время очень часто успех человека зависит от его способности четко мыслить, логически рассуждать и ясно излагать свои мысли.
Цель исследования: повышение интереса школьников к математике как к науке; развитие способности логически рассуждать и приходить к правильным выводам, подробный анализ решения логических задач типа «рыцарь — лжец».
Задачи исследования:
1) изучить основные методы решения логических задач;
2) провести диагностику для определения уровня логического мышления учащихся 5 – 6 классов МБОУ «ЗСОШ №3»;
3) проанализировать результаты диагностики;
4) провести анкетирование учащихся 5-6 классов;
4) подготовить рекомендации для развития логического мышления школьников.
Основные методы (методика): наблюдение, анализ, эксперимент, обобщение экспериментального и теоретического материала.
Подготовка к исследованию
Для начала, мы определили цель исследования – то, что хотим изучить и исследовать. Далее, мы определились с названием исследования, т.е. обозначили тему исследовательской работы. Описали актуальность исследовательской работы, т.е. обосновали выбор именно этой темы работы. Сформулировали цель исследовательской работы и расписали задачи исследовательской работы. Выбрали оптимальный вариант решения проблемы. Составили вместе с учителем план работы для реализации своего исследовательского проекта.
Исторические сведения
Вот что говорили знаменитые ученые. Логика-наука о формах способах мышления. Основы логики были заложены работами учёного и философа Аристотеля (384-322гг. до н.э.). Он пытался найти ответ на вопрос «Как мы рассуждаем?», изучал правила мышления. Аристотель впервые дал систематическое изложение логики. Он подверг анализу человеческое мышление, его формы — понятие, суждение, умозаключение. Так возникла формальная логика.
Мало иметь хороший ум, главное — его хорошо применять. (Р. Декарт). Прежде чем решать задачу-прочитай условие. (Ж. Адамар). Истина ничуть не страдает от того, что кто то ее признает. (Ф. Шиллер).Человека, умеющего наблюдать и анализировать, обмануть просто невозможно. Его выводы будут безошибочны, как теоремы Евклида. (Артур Конан Дойл)
Опираясь на эти высказывания, можно сделать вывод, что задачи на логическое мышление появились еще в глубокой древности, и многие великие ученые придавали большое значение мысли!
Проведение исследования
2.1 Рассмотрение логических задач.
В практической части моей работы мы рассмотрим задачи с тремя типами персонажей — рыцари, лжецы и нормальные люди.
Задачи о рыцарях и лжецах — разновидность увлекательных математических задач, в которых: Лжец (плут, сумасшедший, оборотень) — человек, всегда говорящий ложь. Рыцарь(человек, поступающие правдиво и правильно, правдец) — человек, говорящий всегда правду. Нормальный человек (шпион, человек говорящий, как правду, так и ложь). Решение подобных задач обычно сводится к перебору вариантов с исключением тех, которые приводят к противоречию.
Задача №1
На острове живут рыцари и лжецы. Путешественник, встретивший одного из местных жителей, спросил его, кем он является.
Что ответит житель?
Решение:
Интересно отметить, что и рыцари и лжецы могут произносить фразу «Я — рыцарь». В устах рыцаря это истинное высказывания, лжеца — ложное.
А высказывание «Я — лжец» не может принадлежать ни рыцарю, ни лжецу (т.н. парадокс лжеца).
Задача № 2
По кругу сидят рыцари и лжецы – всего 12 человек. Каждый из них сделал заявление: «Все кроме, быть может, меня и моих соседей – лжецы». Сколько рыцарей сидит за столом, если известно, что лжецы всегда врут, а рыцари всегда говорят правду?
Решение:
Все не могут быть лжецами – тогда все заявления были бы истинными. Значит, есть рыцарь. Все, кроме, быть может, его двух соседей – лжецы. Оба соседа не могут быть лжецами – тогда они сказали бы правду; оба не могут быть рыцарями – тогда бы они солгали. Единственная оставшаяся возможность – один сосед — лжец, другой – рыцарь (то есть два рыцаря рядом, остальные — лжецы) удовлетворяет условиям задачи.
Ответ:2 рыцаря.
Задача №3 :На острове живут 100 рыцарей и 100 лжецов, у каждого из них есть хотя бы один друг. Рыцари всегда говорят правду, а лжецы всегда лгут. Однажды утром каждый житель произнес фразу «Все мои друзья — рыцари», либо «Все мои друзья — лжецы», причем каждую из фраз произнесло ровно 100 человек. Найдите наименьшее возможное число пар друзей, один из которых рыцарь, а другой — лжец.
Решение: Заметим, что в паре рыцарь-лжец каждый должен сказать, что другой лжец: рыцарь скажет правду, а лжец соврёт, в паре рыцарь-рыцарь оба скажут правду, а в паре лжец-лжец оба скажут неправду. Значит фраза «Все мои друзья — лжецы» употребляется только в парах рыцарь-лжец. Минимальное кол-во пар рыцарь-лжец, когда фразу сказали 100 человек, это 50. Если пар будет меньше, то и фраз тоже будет меньше.
Задача №4 : Двое людей A и B, о которых известно, что каждый из них либо рыцарь, либо лжец, либо нормальный человек, высказывают следующие утверждения:
A: B — рыцарь.
B: A — не рыцарь.
Докажите, что по крайней мере один из них говорит правду, но это не рыцарь.
Решение: Эта задача обладает интересной особенностью. Условия ее не позволяют установить, кто из двух островитян говорит правду, не будучи рыцарем: A или B. Мы можем доказать более слабое утверждение: по крайней мере один из двух островитян A и B говорит правду, не будучи рыцарем. Островитянин A либо говорит правду, либо не говорит правду. Докажем два утверждения: 1) если A говорит правду, то он говорит правду, не будучи рыцарем; 2) если A лжет, то B говорит правду, не будучи рыцарем.
1) Предположим, что A говорит правду. Тогда B — рыцарь и, следовательно, говорит правду. Значит, A — не рыцарь. Таким образом, если A говорит правду, то A — лицо, говорящее правду, не будучи рыцарем.
2) Предположим, что A не говорит правду. Тогда B — не рыцарь. Но B должен говорить правду, так как A не может быть рыцарем (ведь A не говорит правду). Следовательно, в этом случае B говорит правду, не будучи рыцарем.
Задача №5
На остров рыцарей и лжецов приехал путешественник и нанял себе проводника. Однажды, увидев вдали туземца, путешественник сказал проводнику: «Пойди и спроси у того человека: рыцарь он или лжец». Вскоре проводник вернулся и сказал: «Этот человек сказал, что он лжец».
Кем был проводник, рыцарем или лжецом?
Ответ: Лжецом
Задача №6
В компании из 12 аборигенов каждый заявил всем остальным: «Вы все лжецы!».
Сколько лжецов в этой компании?
Ответ: 11.
Задача №7
На острове живут 100 человек, причём некоторые из них всегда лгут, а остальные всегда говорят только правду. У каждого жителя острова есть одно любимое время года. Каждому островитянину было задано 4 вопроса:
Любите ли Вы зиму?
Любите ли Вы осень?
Любите ли Вы лето?
Любите ли Вы весну?
На первый и второй вопрос утвердительно ответили по 25 человек, на третий — 45, на четвёртый — 55.
Сколько лжецов на острове?
Решение:
Если бы все жители острова говорили правду, было бы дано 100 утвердительных ответов.
Каждый лжец даёт три утвердительных ответа вместо одного, то есть он увеличивает общее число утвердительных ответов на два.
Так как всего было дано 25 + 25 + 45 + 55=150 утвердительных ответов, то количество лжецов равно 50:2=25.
Задача №8
Возле огрызка Священного Яблока были задержаны 4 аборигена. Агр заявил, что Яблоко съел Бгр, который, в свою очередь, утверждал, что виноват Вгр. Вгр уверял, что Бгр лжёт, а Ггр твердил, что это сделал не он. Выяснилось, что только один из них был рыцарем. Кто рыцарь, и кто обглодал священный фрукт?
Решение:
Бгр и Вгр друг другу противоречат. Значит, один из них рыцарь. Так как рыцарь всего один, то Агр и Ггр лжецы, то есть они врут. Значит Ггр обглодал священное яблоко.
Теперь разберемся кто рыцарь. Так как на виновника мог указать только рыцарь, а на Ггра никто не указал, то рыцарь никого не обвинял, значит рыцарь Вгр.
Задача №9
В стране три города — А, Б, В. Жители города А всегда говорят правду, а города Б — лгут. В городе В лгут и говорят правду в строгой очередности. Дежурному на каланче, увидевшему пожар, позвонили. Состоялся такой разговор: «У нас пожар!» — «Где горит?» — «В городе В!». Куда ехать пожарным?
Ответ: В город А.
Задача №10
Малыш спрятал от Карлсона банку с вареньем в одну из трех разноцветных коробок. На коробках Малыш сделал надписи: на красной – «Здесь варенья нет»; на синей – «Варенье — здесь»; на зеленой – «Варенье в синей коробке». Только одна из надписей правдива. В какой коробке Малыш спрятал варенье?
Ответ: В зеленой.
2.2. Исследование уровня логического мышления учащихся 5-6 классов.
Учащиеся 5-6 классов были разделены на две группы, им было предложено решить два типа задач: логические и математические.
Например:
1 тип (логическая) задача:
Перед нами трое людей A, B и C. Один из них рыцарь, другой лжец и третий — нормальный человек. Эти люди высказывают следующие утверждения.
A: Я нормальный человек.
B: Это правда.
C: Я не нормальный человек.
Кто такие A, B и C?
Решение: Прежде всего заметим, что A не может быть рыцарем, потому что рыцарь не назвал бы себя нормальным человеком. Следовательно получается, что, A — либо лжец, либо нормальный человек. Тогда истинно высказывание человека B. Значит, B — либо рыцарь, либо нормальный человек. Но B не может быть нормальным человеком (так как A — нормальный человек), поэтому B — это доблестный рыцарь, а C — маленький лжец. Но лжец не может сказать о себе, что он не нормальный человек (так как любой лжец — не нормальный человек), и мы приходим к противоречию. Итак, A не может быть нормальным человеком. Следовательно, A — хитрый лжец. Это означает, что высказывание человека B ложно, в силу чего B должен быть нормальным человеком (лжецом он быть не может, так как лжец — человек A). Итак, A — хитрый лжец, а B — нормальный человек. Отсюда мы заключаем, что C — доблестный рыцарь.
2 тип(математическая) задача:
Расстояние по автомобильной дороге от Санкт-Петербурга до Москвы 687 км, от Москвы до Ростова-на-Дону 1064 км, от Ростова на Дону до Сочи 712. Сколько километров нужно проехать на автомобиле, чтобы из Санкт-Петербурга приехать в Сочи через Москву и Ростов на Дону?
Решение:
687+1064=1751((км)от С-П до Р-Д)
1751+712=2463((км)от С-П до Сочи)
Ответ: От Санкт-Петербурга до Сочи 2463 километра.
Из диаграммы видно, что 13 учащихся (43%) успешно решили обе задачи, с математической задачей – 18 учащихся (60%), со второй задачей на логику справились лишь 9 учеников (30%) и не справились с решением задач 3 учащихся (10%)
Подводя итог, можно сделать вывод, что с задачами более простыми в целом ученики 5-го и 6-го классов справляются, но если добавляются немного больше элементов в рассуждениях то справляются с такими заданиями не все.
Анализируя полученные результаты, в целом можно сказать, что лучше с решением логических задач справились те учащиеся, которые встречали подобные задачи при подготовке к олимпиаде или на факультативах. Ученики, занимающиеся лишь в рамках школьной программы, показали хуже результаты, возможно причиной этому является, что для решения данного вида задач требуется хорошее знание математики, ученики 5х классов пока ещё не имеют опыта в решении таких задач.
2.3 Социальный опрос
Также мы провели соц. опрос среди учащихся 5-6 классов. Всем задали вопрос: «Какие задачи легче решать: математические или логические? В опросе участвовали 25 человек. 15 человек ответили – математические, 6-логические, 4 — никакие не смогут решить. Результат опроса представлен на диаграмме:
Выводы
Анализируя полученные в ходе исследований результаты, в целом можно сказать, что лучше с решением логических задач справились те учащиеся, которые встречали подобные задачи при подготовке к олимпиаде или на факультативах. Ученики, занимающиеся лишь в рамках школьной программы, показали хуже результаты, возможно причиной этому является, что для решения данного вида задач требуется хорошее знание математики, ученики 5х классов пока ещё не имеют опыта в решении таких задач.
Заключение
В данной работе Вы познакомились с логическими задачами. С тем, что такое логика. Вашему вниманию были предложены различные логические задачи, которые помогают развивать логическое и образное мышление.
У любого ученика есть стремление к познанию, желание проверить себя. Чаще всего способности школьников так и остаются не раскрыты для них самих, они не уверены в своих силах, равнодушны к математике.
Для таких школьников, мы предлагаем применять логические задачи. Эти задачи могут быть рассмотрены на кружковых и факультативных занятиях. Для этого мы создали небольшой сборник логических задач, который можно использовать при подготовке к олимпиадам, на факультативах и для развития логического мышления учеников. Они должны быть доступны, будить сообразительность, овладевать их вниманием, удивлять, пробуждать их к активной фантазии и самостоятельному решению.
Также мы считаем, что логика помогает нам в нашей жизни справиться с любыми трудностями, и все что мы делаем, должно быть логически осмысленно и построено. С логикой и логическими задачами мы сталкиваемся не только в школе на уроках математики, но и на других предметах.
Список литературы
1. Рэймонд М. Смаллиан. Как же называется эта книга? – М.: АСТ, 2013
Логика и рассуждения. Лжецы и рыцари. Логические задачи, головоломки, тесты на интеллект, логические игры
Лжецы и рыцари
На острове живут 100 рыцарей и 100 лжецов, у каждого из них есть хотя бы один друг. Рыцари всегда говорят правду, а лжецы всегда лгут. Однажды утром каждый житель произнес фразу «Все мои друзья — рыцари», либо «Все мои друзья — лжецы», причем каждую из фраз произнесло ровно 100 человек. Найдите наименьшее возможное число пар друзей, один из которых рыцарь, а другой — лжец.
Ответ
: Заметим, что в паре рыцарь-лжец каждый должен сказать, что другой лжец: рыцарь скажет правду, а лжец соврёт, в паре рыцарь-рыцарь оба скажут правду, а в паре лжец-лжец оба скажут неправду. Значит фраза «Все мои друзья — лжецы» употребляется только в парах рыцарь-лжец. Минимальное кол-во пар рыцарь-лжец, когда фразу сказали 100 человек, это 50. Если пар будет меньше, то и фраз тоже будет меньше.
Рейтинг:
+269
Комментарии:
Витя, 2010-04-27
Хорошая задачка нормально
ERUDIT, 2010-04-27
ой-ой-ой, HeeL... А что уже неоткуда задачки брать?
/forum/index.php/topic,2653.0.html
ERUDIT, ну почему же нету - есть, форум полон задач: http://nazva.net/forum/index.php
potato, 2010-04-29
"у каждого из них есть хотя бы один друг"... а если друзей больше 1? И допустим среди них есть и рыцари и лжецы,тогда лжец может сказать любую фразу
Никита, 2010-05-04
50
Vik, 2010-05-05
2 100 минимальное количество, а10000 - максимальное. В задачи не было условия о том, что рыцарь может дружить только с одним лжецом и наоборот, а если лжецы дружат между собой, а рыцари дружат только с лжецами? л-Л--Р-л р-Л--Л-р р-Р--Р-р
Вот если схематично - 3 вида пар. Сказать мой друг лжец может только кто либо из пары Л-Р. а это сказало 100 человек, следовательно 50 пар. остальные добираются связками ЛЛ, РР в любой пропорции.
Естественно при условии что у 1 человека может быть только 1 друг. в условии этого явно не сказано, но думаю что все таки подразумевалось в вопросе.
интер съедается поэтому схемка вышла неявной. В общем 3 пары ЛЛ (оба скажут Р) РР (оба скажут Р) ЛР (оба скажут Л)
игорь, 2010-08-15
ни одной 125 О_о фигня, хехе!!!
Даша, 2011-01-29
ни одной
pasa, 2011-02-17
ни одной?
то есть рыцари дружат только с рыцарями, а лжецы - только с лжецами?
кто же тогда сказал фразу "Все мои друзья — лжецы"?
правильный ответ 50
Filifionka, 2011-03-14
ЗдОрово! Только перечитав снова задачу, наконец сообразила, почему пар не может быть меньше 50. Фразу "...причем каждую из фраз произнесло ровно 100 человек" я изначально поняла так, что общее количество людей, которые произнесли первую или вторую фразу, равняется 100 из 200. Не обратила внимания на слова "каждую из фраз". Таким образом, было достаточно просто выяснить, что фразу "Все мои друзья - лжецы" произносят только в парах Рыцарь-Лжец. Всех людей, произнесших эту фразу, 100, а пар соответственно 50. Вот что значит невнимательность... Лжецы врали, когда сказали: "Все мои друзья — рыцари". Поэтому я думала, что из 3ех видов возможных пар правдив только "рыцарь-рыцарь". Мой ответ - 0. элементарно Вася!
Еф, 2011-08-05
Стандартная задача оценка+пример. Примера в ответе нет, хотя б ради приличия написали бы, что он очевиден.
semen063, 2011-10-24
Рыцарь - "Все "лжецы"-лжецы"
Лжец - "Все рыцари говорят правду")))) Парадокс почему 50 если 100 рыцарей и 100 полуцается 100. 100 рыцарей сказали правду 100 лжецов соврали то есть при помощи математики 100+100=200/2=100
Дево4ка, 2012-08-27
50= 100:2
Дядя Женя, 2012-11-04
так как по условию не оговоренно, что все дружат только парами => 1 рыцарь может дружить со всеми лжецами, и условие будет соблюдено... 99 рыцарей скажут, что у них в друзьях только рыцари, + 1 лжец соврёт, что у него в друзьях только рыцари. В свою очередь 1 рыцарь скажет правду, что у него в друзьях только лжецы, и 99 лжецов солгут, что у них в друзьях только лжецы!.. ИМХО
Елена, 2013-08-10
100 рыцарей могут быть и 100 лжецами
Макс , 2013-12-14
100
александр, 2014-05-16
минимальное число пар рыцарь-лжец это 1. остальные 198 чел, из которых 96 рыцари и 96 лгуны, могут объединиться в 48 пар из одних и 48 пар других, не зависимо от того, что они там утром с бодуна сказанули. Это не противоречит условию. Следуя логике, таких пар может быть МИНИМУМ 1, а даже не 50) 2
"Заметим, что в паре рыцарь-лжец каждый должен сказать, что другой лжец: рыцарь скажет правду, а лжец соврёт" -разве Лжец скажет при знакомстве что он Лжец???, для каждого Рыцаря друг Рыцарь, будь его друг Рыцарь или Лжец!!!!!!!!!!!!
Михаил, 2016-03-18
Ответ задачи не верный. В задаче явно указано, что друзей может быть несколько ("хотя бы один" означает >0), т.е. совсем не обязательно разбивать всех на пары. Правильный ответ 1. Только один рыцарь дружит со лжецом, но этот лжец дружит, ещё со всеми остальными лжецами. 99 рыцарей и этот один лжец говорят фразу "Все мои друзья рыцари". 99 лжецов и этот один рыцарь говорят фразу " Все мои друзья лжецы".
Михаил, 2016-03-18
Поправка,к предыдущему комментарию. Т.к. лжецы обязательно должны говорить неправду, пусть все лжецы, например, дружат с этим одним рыцарем и,каждый из них дружит еще хотя бы с одним лжецом. Тогда, они могут говорить любую из двух фраз, любая будет неправдой. Ответ: 1.
Михаил, 2016-03-18
Извиняюсь, ошибка все-таки в моих двух комментариях выше, а не в ответе к задаче. Первый мой комментарий не верен, т.к. лжецы обязательно должны говорить неправду, второй - т.к. если 99 лжецов начнут дружить с рыцарем, кол-во пар Л-Р тоже увеличется на 99. Минимальное кол-во пар действительно 50. К этому числу, как альтернативный вариант, можно прийти пытаясь максимизировать число рыцарей, которые дружат только с рыцарями, и число лжецов, которые дружат только с лжецами, и те и другие должны будут сказать фразу "Все мои друзья рыцари", а т.к. максимальная их сумма равна 100, логично в каждую из этих групп включить по 50 чел. Все оставшиеся рыцари и лжецы (их тоже по 50), вынуждены составить пары Р-Л,т.к. они должны будут сказать фразу "Все мои друзья лжецы". А мне не нравится слово ВСЕ в фразах "Все мои друзья - лжецы" и "Все мои друзья - рыцари". И вообще-то и сами фразы. Если среди друзей рыцаря, который говорит правду, есть и рыцари, и лжецы? Как он может сказать, не солгав,"Все мои друзья - лжецы" или "Все мои друзья - рыцари"? Не понятно... Неувязка...
(PDF) Кванторы на Острове Рыцарей и Лжецов / Quantifiers on the Island of Knights and Knaves
6. Допустим, что A — рыцарь. Тогда, согласно его заявлению,
все, кроме него, — лжецы. В частности, лжецом является и B.
Значит, высказывание B ложно. Но B (в других словах) утвер-
ждал, что A говорит правду. Поскольку это оказалось неверным,
A солгал, что для рыцаря невозможно. Полученное противоре-
чие показывает, что A — не рыцарь, то есть — лжец. Поскольку
B утверждал, что A говорит правду, он тоже — лжец.
Далее, поскольку высказывание, сделанное A, ложно, оказы-
вается неверным, что все жители Острова, отличные от A, явля-
ются лжецами. Иначе говоря, не все жители Острова, отличные
от A, являются лжецами. Значит, существуют жители, отличные
от A и не являющиеся лжецами. Ясно, что они являются рыца-
рями. Итак, A и B оба — лжецы, но есть и рыцари.
7. Сначала заметим, что высказывание, произнесённое A, явля-
ется примером импликации, т. е. предложением вида “если ТАК,
то ЭТАК”, где на месте “ТАК” и “ЭТАК” стоят какие-нибудь
предложения. Предложение, стоящее на месте “ТАК”, называет-
ся посылкой импликации, предложение, стоящее на месте
“ЭТАК” — заключением. Импликация считается ложной, когда
посылка истинна, а заключение — ложно, и считается истинной во
всех остальных случаях. Обратимся к задаче.
Допустим, что A — лжец. Тогда произнесённая им имплика-
ция ложна. Значит, ложно и заключение этой импликации, в ко-
тором утверждается, что A — лжец. Оно, однако, должно быть
истинным (поскольку A, по предположению, лжец). Полученное
противоречие показывает, что A — не лжец, то есть рыцарь.
Теперь, поскольку A — рыцарь, импликация, произнесённая
им, истинна, а заключение ложно. Значит, и посылка должна
быть ложной. Иначе говоря, неверно, что в описанной группе
жителей Острова лжецов нет. Раз это неверно, лжецы в ней есть.
Теперь ясно, что рыцарей в группе быть не могло, ибо, согласно
условию, все члены группы утверждали, что лжецов среди них
нет, т. е. произносили ложное высказывание, что для рыцарей
невозможно. Итак, A был рыцарем, а описанная группа жителей
Острова состояла из одних лжецов.
Два сундука. Лжец и Рыцарь — Логические задачи — Занимательная математика — Каталог статей
(Ф. Хаусдорф.)
‘
quotes[1]='»Математика — это язык, на котором написана книга природы.»
(Г. Галилей)
‘
quotes[2]='»Кто с детских лет занимается математикой, тот развивает внимание, тренирует свой мозг, свою волю, воспитывает настойчивость и упорство в достижении цели.»
(А. Маркушевич)
‘
quotes[3]='»Рано или поздно всякая правильная математическая идея находит применение в том или ином деле.»
(А.Н. Крылов)
‘ quotes[4]='»Если вы хотите участвовать в большой жизни, то наполняйте свою голову математикой. Она окажет вам потом огромную помощь во всей вашей работе.»
(М.И. Калинин)
‘
quotes[5]='»Разве ты не заметил, что способный к математике изощрен во всех науках в природе?»
(Платон)
‘
quotes[6]='»Математика есть лучшее и даже единственное введение в изучение природы.»
(Д.И. Писарев)
‘
quotes[7]='»Вдохновение нужно в геометрии не меньше, чем в поэзии.»
(А.С. Пушкин)
‘
quotes[8]='»Геометрия полна приключений, потому что за каждой задачей скрывается приключение мысли. Решить задачу – это значит пережить приключение.»
(В. Произволов)
‘
quotes[9]='»В математике есть своя красота, как в живописи и поэзии.»
(Н.Е. Жуковский)
‘
quotes[10]='»Химия – правая рука физики, математика – ее глаз.»
(М.В. Ломоносов)
‘
quotes[11]='»Математику уже затем учить надо, что она ум в порядок приводит.»
(М.В. Ломоносов)
‘
quotes[12]='»Математика — это язык, на котором говорят все точные науки.»
(Н.И. Лобачевский)
‘
quotes[13]='»Именно математика дает надежнейшие правила: кто им следует – тому не опасен обман чувств.»
(Л. Эйлер)
‘
quotes[14]='»Числа не управляют миром, но они показывают, как управляется мир.»
(И. Гете)
‘
quotes[15]='»Было бы легче остановить Солнце, легче было сдвинуть Землю, чем уменьшить сумму углов в треугольнике или свести параллели к схождению…»
(В.Ф. Каган)
‘
quotes[16]='»Счет и вычисления — основа порядка в голове.»
(Песталоцци)
‘
quotes[17]='»Величие человека — в его способности мыслить.»
(Б. Паскаль)
‘
quotes[18]='»Если вы хотите научиться плавать, то смело входите в воду, а если хотите научиться решать задачи, то решайте их.»
(Д.Пойа)
‘
quotes[19]='»Предмет математики столь серьезен, что не следует упускать ни одной возможности сделать его более занимательным.»
(Б. Паскаль)
‘
quotes[20]='»В математических вопросах нельзя пренебрегать даже самыми мелкими ошибками.»
(И. Ньютон)
‘
quotes[21]='»Первое условие, которое надлежит выполнять в математике, — это быть точным, второе — быть ясным и, насколько можно, простым.»
(Л. Карно)
‘
quotes[22]='»Много из математики не остается в памяти, но когда поймешь ее, тогда легко при случае вспомнить забытое.»
(М.В. Остроградский)
‘
quotes[23]='»Математика — это цепь понятий: выпадет одно звенышко — и не понятно будет дальнейшее.»
(Н.К. Крупская)
‘
quotes[24]='»Математика уступает свои крепости лишь сильным и смелым.»
(А.П. Конфорович)
‘
quotes[25]='»Доказательство — это рассуждение, которое убеждает.»
(Ю.А. Шиханович)
‘
quotes[26]='»В каждой естественной науке заключено столько истины, сколько в ней есть математики.»
(И. Кант)
‘
var whichquote= Math.floor(Math.random()*(quotes.length))
document.write(quotes[whichquote])
Рыцари и лжецы — ЕвРоПа-кидс
Е. М. Шамахова
Здравствуйте, мои маленькие читатели и мыслители!
Кстати, вы знакомы с МатеМашей и ПрограМишей? Знакомьтесь, это мои хорошие друзья, они очень интересные и любознательные ребята! Они все время задают мне разные вопросы. И потом мы вместе с ними начинаем искать… Что? Как что? Конечно же, ответы! И где мы только не путешествуем в поисках ответов, чего только интересного не находим по дороге! Кто-то звонит в дверь… Наверно, это они, МатеМаша и ПрограМиша. — Евгения Михайловна, здравствуйте! – прямо с порога, не раздевшись, кричит ПрограМиша, — мы сегодня в школе по пятерке получили! — Молодцы! А я вам тоже приятный сюрприз приготовила. — Какой? — Сегодня мы отправимся путешествовать! — Здорово! А что, задачки решать не будем? — Как это не будем? Будем! Мы же будем путешествовать по океану! Но не простому, а Математическому. Собирайтесь! — А что брать с собой? – спросила очень аккуратная и практичная МатеМаша. — Берем только самое необходимое: бумагу и ручку. Да, и самое главное не забыли? — А что самое главное? — Конечно, голова! Без нее мы точно не справимся! — Голова на месте! – ПрограМиша даже снял свою любимую панамку, чтобы показать, что самое главное он не забыл. — Ну, тогда полный вперед! – скомандовала я, — сегодня мы поплывем на очень известный математический остров, остров Рыцарей и Лжецов. Все математики, особенно маленькие, очень любят приезжать в гости на этот остров. — А почему он так называется? – любопытному ПрограМише всегда хотелось выяснить все подробности. — Пока мы плывем на остров, я немножко о нем расскажу. Остров этот необычный, живут на нем рыцари и лжецы. Рыцари всегда говорят только правду, а лжецы всегда только лгут. — Ах, нехорошие врунишки! – не сдержалась МатеМаша. — Не расстраивайся, — утешила я МатеМашу, — нам, математикам, это не помеха, мы даже из неправды можем узнать много полезного! Ну, вот мы и на месте. Смотрите, первый местный житель. — Но как же мы у него узнаем, рыцарь он или лжец? Если просто спросить его, он ведь может и соврать, — задумалась МатеМаша. — Вот вам и первая задача:
Задача 1. Что ответит житель этого острова, если его спросить: «Ты рыцарь?»
— Да, непростая задача, — поддержал ПрограМиша, — но, кажется, я знаю ответ. И этот ответ не дает нам никакой информации… — Точно, — согласилась МатеМаша, — если он и правда рыцарь, он честно ответит «да», а если это лжец, то честно было бы сказать «нет», но он всегда говорит неправду, поэтому тоже скажет «да». Получается, что ответ будет в любом случае «да», так что нечего и спрашивать. — Молодцы, с первой задачей вы справились. Посмотрите, вот идет еще один местный житель. Кажется, они приятели. Давайте с ними пообщаемся. И мы пошли разговаривать с двумя жителями. А вам, мои любезные читатели, даем еще задачу, которую вы попробуйте решить сами:
Задача 2. Вот что сказали нам эти два жителя острова в разговоре. Первый: «Мой приятель лжец». Второй: «Нет, мы оба рыцари». Кем же является каждый из них на самом деле?
— Кажется, — не унимался ПрограМиша, — у меня уже есть идея! Хорошо, что эти жители оказались такими дружелюбными и разговорчивыми. — Подожди, ПрограМиша, не говори ответ, дай нашим читателям тоже подумать над этой задачкой! А вот вам пока еще одна задача, потруднее:
Задача 3. Как-то раз встретила я двух жителей этого острова и спросила, кто они. Первый ответил мне: «Хотя бы один из нас лжец». А второй ничего не сказал. Попробуйте определить, кто из них кем является.
— Как, второй совсем-совсем ничего не сказал? А как же мы узнаем, кто он? — Вот и подумайте! А сейчас пора по домам. — До свидания, Евгения Михайловна! Мы обязательно все решим! — Даже не сомневаюсь! Пока, мои юные математики! До новой встречи!
Ответы и решения к задачам 2 и 3:
Задача 2. Оба жителя рыцарями быть не могут, поэтому второй житель точно сказал неправду. Значит, он лжец. Значит, слова первого жителя – правда, и он рыцарь.
Задача 3. Говоривший житель сам не может быть лжецом (иначе его слова правда, хотя бы один лжец среди есть – он сам). Значит, он рыцарь. Но чтобы его слова были правдой, среди них должен быть и лжец – это второй житель, который промолчал.
Копирование статьи или её частей разрешается только с указанием автора и ссылкой на сайт https://europa-kids.com
Назад в Числоград
Простая задача рыцарей и лжецов в SWI-Prolog
Я пытаюсь решить проблему «Knights and Knaves», используя метод generate-and-test для N человек, чтобы достичь следующего результата:
Вышеприведенный результат можно объяснить следующим образом:
Людей столько же, сколько целых чисел в первом списке.
Каждый человек заявляет: «по крайней мере, X из нас-лжецы» , причем X -это число в первом списке, которое соответствует каждому человеку.
Люди, обозначенные 1 в списке Knaves , оказались лжецами.
Мой подход:
Моя мысль состояла в том, чтобы создать все перестановки списка Knaves , содержащие 0 и 1 , а затем отфильтровать те, которые не соответствуют требованиям людей:
Вышеприведенный код создает все перестановки, но теперь я застрял в том, как мне следует действовать, сохраняя только одну правильную перестановку.
Я уверен, что должна быть рекурсия, где я должен пройти через список Statements , но тогда я не уверен, каким должно быть условие. Я думаю что-то вроде следующих строк (псевдокод):
check([Head|Tail], Results) :-
% Condition:
% Head three or more times in the Results
check(Tail, Results).
Движется ли мой подход в правильном направлении? Если нет, то как лучше всего решить эту проблему?
list
prolog Поделиться Источник Angel Politis14 мая 2019 в 21:01
1 ответ
Python интерфейс с SWI-Prolog
Я хочу использовать скрипт Python в качестве интерфейса для программы Prolog, которая использует движок SWI-PL. Итак, компонентами установки являются: Python (2.7 или выше) SWI-PL: сайт здесь Я искал интерфейс между SWI-PL и Python. То, что я нашел, это: PySwip но это, по-видимому, отсутствует из…
Использование предиката вычитания в swi prolog
Мне нужно вычесть определенные числа из данного списка. Я использую SWI Prolog. Вот что я сделал. subtract([1,4],[1,2,3,4,5],’L’) Но, похоже, работа в SWI prolog..pls мне не помогает….
4
Вот минимальное дополнение к вашему решению (слегка переименованные некоторые предикаты):
Вы могли бы использовать same_length/2 вместо length/2 . Вы также можете заменить рекурсивный предикат на maplist . Код становится:
statements_knaves(S, K) :-
same_length(S, K),
maplist(knave, K),
sum_list(K, N),
maplist(validate(N), S, K, _).
knave(0).
knave(1).
validate(N, X, 0, knight) :- X =< N.
validate(N, X, 1, knave) :- X > N.
Поделиться User921315 мая 2019 в 10:50
Похожие вопросы:
Prolog определение типа в swi-prolog
в visual prolog есть раздел domains в программе prolog, в котором вы можете определять типы. Есть ли что-то подобное в swi-prolog? В visual prolog тип определяется следующим образом: domains NewType…
Interprolog и SWI-Prolog
Я настроил и установил как interprolog, так и swi prolog на своей машине linux, следуя инструкциям здесь : Interprolog with SWI instructions on Linux Я отредактировал unixVariables.sh, чтобы он…
Унификация динамических предикатов в SWI Prolog
Я вызываю движок SWI Prolog из C++ dll и хотел бы иметь доступ ко всем утвержденным/динамическим предикатам (аналогично тому, что обычно делает листинг). В GNU Prolog я бы назвал предикат dynamic/1…
Python интерфейс с SWI-Prolog
Я хочу использовать скрипт Python в качестве интерфейса для программы Prolog, которая использует движок SWI-PL. Итак, компонентами установки являются: Python (2.7 или выше) SWI-PL: сайт здесь Я…
Использование предиката вычитания в swi prolog
Мне нужно вычесть определенные числа из данного списка. Я использую SWI Prolog. Вот что я сделал. subtract([1,4],[1,2,3,4,5],’L’) Но, похоже, работа в SWI prolog..pls мне не помогает….
Неопределенная ошибка процедуры в prolog SWI
Запуск prolog SWI на windows 8 в первый раз. это моя программа (.pl) файл, очень простой только с 3 фактами: (я полный новичок prolog) hello. a. b. Когда я загружаю его (консультируюсь) в prolog-SWI…
Получите несколько решений в SWI-Prolog
Я новичок в SWI-Prolog (но имею некоторый опыт работы в Borland Prolog), и я столкнулся со странным поведением для следующего тестового кода: test(10). test(1). Ожидается, что запрос ?-test(A)…
в чем разница между #= и =:= в SWI prolog
В чем разница между #= и =:= в SWI prolog. Я нашел определение из SWI prolog, но все еще путаюсь в нем. http://www.swi-prolog.org/pldoc/man? section=arithpreds http://www.swi-prolog.org/pldoc/man?…
Prolog: печать на консоль swi prolog сразу же я консультируюсь с файлом
У меня есть простая программа prolog: write_manual:- write(‘——————————‘), write(‘USAGE MANUAL’), write(‘find_course. — List all the available courses’),…
приоритет отрицания в SWI prolog
Вот цитата из книги Блэкберна и Бос Representation and Inference for Natural Language. :- op(900,yfx,>). % implication :- op(850,yfx,v). % disjunction :- op(800,yfx,&). % conjunction :-…
Логика
— Рыцари и лжецы: кто такие B и C? (задание 26 из «Как называется эта книга?»)
У меня следующий выпуск № 26 из Как называется эта книга? Р. Смулляна:
Пазлы самые разные.
об острове, на котором
жители называли «рыцарей» всегда
сказать правду, и другие звонили
«негодяи» всегда лгут. Предполагается
что каждый житель острова
либо рыцарь, либо лжец.мне нужно
начнем с хорошо известной загадки этого
введите, а затем введите
множество собственных головоломок.
Согласно этой старой задаче три
жителей — A, B и C —
стояли вместе в саду. А
незнакомец прошел мимо и спросил А: »
ты рыцарь или лжец? »А ответил:
но довольно нечетко, поэтому
незнакомец не мог разобрать, что он
сказал. Незнакомец спросил Б: «Что
сказал А? «Б ответил:» А сказал, что он
лжец «. На этом этапе третий
человек, C, сказал: «Не верьте B; он
врут! »Вопрос в том, какие Б
и C?
Я предположил, что можно использовать таблицы истинности, и составил следующее:
Мы используем $ A $, когда A — рыцарь, и
$ \ neg A $, когда A — лжец.
$ F1 $ — это то, что сказал B ($ A \ leftrightarrow \ neg A $),
я. е. B сказал, что A сказал, что он лжец.
Следовательно, B говорит правду, если
и только если он рыцарь ($ B $).
$ F2 $ означает, что C — рыцарь, если и
только если он говорит правду, т. е.
B — лжец ($ \ neg B $).
$ G $ позволяет нам выбирать только те
претензии между $ F1 $ и $ F2 $
которые верны.
Могу я с уверенностью сказать, что у нас всего два случая, когда $ G $ истинно и можно сделать следующие выводы:
B — лжец, потому что в соответствующих строках есть $ 0 $ s (false).
C — рыцарь, потому что B лжет, и в соответствующих строках есть $ 1 $ s (true).
Мы не можем точно сказать, что такое A, потому что мы не могли разобрать, что он сказал, и в таблице есть два случая с $ 0 $ и $ 1 $ в соответствующих строках, где $ G $ истинно.
Скажите, пожалуйста, верны ли мои расчеты и таблица истинности, а не только вывод? Лучший ответ — это тот, который либо объясняет, чего мне не хватает в моей таблице истинности, либо содержит правильный ответ вместо моего, предположительно ошибочный.Я пытаюсь понять, как их можно использовать, и я полагаю, что с этой проблемой довольно просто поиграть, в конце концов, у вас в голове те же рассуждения.
Заранее спасибо.
15-110: Принципы вычислений
15-110: Принципы вычислений
15-110 Весна 2011 г. Класс
Примечания: Knights and Knaves (Выполнимость)
Основы
Рыцари: Всегда говори правду.
Кнейвс: Всегда лгите.
Задача:
Дано: Одно утверждение, каждое из N символов, каждое рыцарь или лжец.
Задача: определить, кто рыцарь, а кто лжец.
Примеры задач:
Knights-and-Knaves как выполнимость
Начните с задачи Knights-and-Knaves.
Например, вот задача № 1 из Хранилища Рыцарей и Кнейвов: «Очень особенный остров населен только рыцарями и лжецами.Рыцари
всегда говорят правду, а мошенники всегда лгут. Вы встречаете двух жителей: Зои и Мела. Зои говорит вам, что Мел — лжец. Мел говорит: «Ни Зои, ни я не лжецы». Итак, кто такой рыцарь, а кто лжец? »
Присвойте (заглавную) букву каждому персонажу.
Здесь мы назначим Z для Зои, а M для Мел.
Пусть A означает« A есть рыцарь ». Таким образом,« не А »означает« А не рыцарь »(то есть« А — лжец »).
Таким образом, Z означает« Зоя — рыцарь », а М означает« Мел — это рыцарь ». рыцарь.
Пусть SA будет утверждением A, представленным в виде логического выражения. Сделайте это для каждого символа.
Зои говорит: «Мел — валет», поэтому SZ = (не M)
Мел говорит: «Ни одна Зои и я не лжец », поэтому SM = (Z и M)
Рассмотрим:
Мы знаем эту тавтологию:« A — рыцарь »или« A — не рыцарь »
Мы можем переписать эту тавтологию в виде логического логика как: A или (не A)
Теперь, если «A — рыцарь», то SA тоже должно быть истинным (поскольку A никогда не лжет).Таким образом, A эквивалентно (A и SA).
Аналогично,
если «A — лжец», то SA должно быть ложным (поскольку A всегда лжет).
Таким образом (не A) эквивалентно ((не A) и (не SA)).
Если мы сделаем два предыдущих изменения в A или (не A) с шага 2, мы получим: … (A и SA) или ((не A) и (не SA))
This
это общая информация, которую мы получаем из утверждения персонажа A.
Назовите это IA (для информации из заявления A). Итак: IA = (A и SA) или ((не A) и (не SA)) Вы могли заметить, что это то же самое, что сказать: IA = (A = SA)
Продолжая конкретный пример :…
IZ = (Z = SZ)
IM = (M = SM)
Наконец,
чтобы решить загадку, мы требуем, чтобы информация, которую мы черпаем из
утверждения каждого персонажа верны одновременно. Если мы
вызовите решение головоломки PS, тогда PS = IA и IB и … и
IQ (столько персонажей в головоломке).
Итак, в примере PS = IZ и IM
Теперь мы просто используем таблицы истинности, чтобы найти присвоение истинности переменным, которое делает PS истинным.(Это выполнимость!)
Итак, в примере:
M
Z
SM = (Z и M)
SZ = (не M)
IM = (M = SM)
IZ = (Z = SZ)
ПС = ИЗ и ИМ
0
0
0
1
1
0
0
0
1
0
1
1
1
1
1
0
0
0
0
1
0
1
1
1
0
1
0
0
Итак, ответ: Мел — Валет. Зоя — рыцарь.
Давайте проверим нашу работу:
«Зои говорит вам, что Мел — лжец» Это правда (Мел — лжец), чего мы и ожидаем, поскольку Зоя — рыцарь. Проверять!
«Мэл говорит:« Ни Зои, ни я не лжецы ». Поскольку Зоя рыцарь, это ложь. Но Мел — Валет, поэтому ожидается, что он солгает. Проверять!
Другой пример
Начните с задачи рыцарей и ловушек.
Например, вот задача № 51 из хранилища рыцарей и лжецов: «Особый остров населен только рыцарями и лжецами.Рыцари
всегда говорят правду, а негодяи всегда лгут. Вы встречаете трех жителей: Алису, Рекса и Боба. Алиса говорит тебе
что Рекс — лжец. Рекс говорит вам, что Боб лжец — это неправда.
Боб заявляет: «Я рыцарь или Алиса — рыцарь». Итак, кто такой рыцарь, а кто лжец? »
Назначьте (заглавную) букву каждому персонажу.
Здесь мы назначим A для Алисы, B для Боба и R для Рекса.
Пусть Означает «А — рыцарь». И поэтому «не А» означает «А не рыцарь» (то есть «А — лжец»).
Таким образом, A означает «Алиса — рыцарь», B означает «Боб — рыцарь», а R означает «Рекс — рыцарь».
Пусть SA будет утверждением A, представленным в виде логического выражения. Сделайте это для каждого персонажа.
Алиса говорит: «Рекс — лжец», поэтому SA = (не R)
Боб говорит: «Я рыцарь или Алиса — рыцарь», поэтому SB = (B или A)
Рекс говорит: «Неверно, что Боб лжец», поэтому SR = B. [или, если быть более точным, (не (не B))]
Мы назначаем IA, IB и IR следующим образом:
IA = (A = SA)
IB = (B = SB)
IR = (R = SR)
Мы назначаем PS следующим образом:
Итак, в примере PS = IA и IB и IR
Теперь мы просто используем таблицы истинности, чтобы найти присвоение истинности переменным, которое делает PS истинным.(Это выполнимость!)
«Алиса говорит вам
что Рекс — лжец ». Это ложь (Мэл не лжец), чего мы и ожидаем, поскольку Алиса — лжец. Проверьте!
« Рекс говорит вам, что это ложь, что Боб лжец. « Это
правда (так как Боб рыцарь, неверно, что он лжец),
чего мы и ожидаем, так как Рекс рыцарь. Проверять!
«Боб заявляет:` Я рыцарь или Алиса рыцарь.'» Это
правда (поскольку Боб — рыцарь, поэтому не имеет значения, что Алиса — лжец),
чего мы и ожидаем, поскольку Боб рыцарь. Проверять!
Роберт Л. Холлидей — Решения и другие проблемы
Как и было обещано, вы найдете решения головоломок Смоллиана и другие варианты головоломок из статьи Лжецы и правдивые: изучение логики от Раймонда Смоллиана , появившейся в Ноябрьский (2005 г.) выпуск Math Horizons .
Knights and Knaves — Стандартное тарифное решение
(I) Предположим, А действительно сказал, что среди трех был (ровно) один рыцарь. Тогда B должен быть рыцарем, чтобы точно передать то, что сказал A. Поскольку C не согласен с B, C должен быть лжецом. Итак, если A — лжец, у нас был бы лжец A, делающий истинное утверждение. Если A — рыцарь, тогда в группе будет два рыцаря, поэтому A никогда не сделает такого заявления. Таким образом, независимо от того, к какому типу принадлежит А, он не может утверждать, что среди них есть один рыцарь.Значит, B должен лгать, а C говорит правду. Опять же, мы не знаем, что за человек А.
(II) Говорящий не может быть рыцарем, утверждающим, что он лжец. Значит, оратор — лжец. Если бы друг был рыцарем (как утверждает оратор), то у нас был бы лжец, делающий правдивое заявление. Так что друг тоже лжец.
Добавление нормалей в раствор смеси
(I) Предположим, что A нормальный. Тогда A говорит правду, B соглашается, поэтому B также говорит правду и, следовательно, является рыцарем.Это делает Си лжецом — к сожалению, лжецом, который говорит правду.
Предположим, A рыцарь. Тогда A лжет!
Таким образом, мы пришли к выводу, что A — лжец. Поскольку лжецы лгут и B соглашается, то B также лжет. Следовательно, B не может быть рыцарем, поэтому должен быть нормальным. C — рыцарь.
(II) Если A рыцарь, то B должен быть нормальным, а C — лжецом. Итак, C солгает и скажет, что B имеет более высокий ранг, чем A.
Если A лжец (и лжец), мы можем заключить, что C — рыцарь, а B — нормальный.C скажет правду и скажет, что B имеет более высокий ранг, чем A.
Если A нормальный и говорит правду, то B — рыцарь, а C — лжец. Но теперь у нас есть противоречие, а именно, рыцарь (B) лжет.
Если A нормальный и лжет, то C — рыцарь, а B — лжец. Снова мы имеем противоречие, на этот раз лжец (B) говорит правду.
Итак, на самом деле A не является нормальным, и только в других возможных случаях ответ C будет: «B имеет более высокий ранг, чем A.«
The Planet Og Solutions
(I) Один вопрос, который определяет цвет: Вы с Севера? Чтобы увидеть это, нам просто нужно подумать, как отреагирует каждый из четырех типов жителей:
зеленый северянин — говорит правду и говорит «Да». зеленый южанин — врет и говорит «да». красный северянин — врет и говорит «Нет». красный южанин — говорит правду и говорит «Нет».
Таким образом, ответ «Да» идентифицирует говорящего как зеленый, а ответ «Нет» идентифицирует говорящего как красный.
(II) Эксперт по логике, должно быть, разговаривал с зеленым жителем. Если бы эксперт разговаривал с красным жителем, так как было средь бела дня, он бы знал, что житель лжет. Если бы обитатель был красным и лгал, логик мог бы сделать вывод, что говорящий был красным северянином. Следовательно, логик обращался к зеленому жителю.
(III) Предположим, что A зеленый. Тогда B красный и тоже лжет. Значит, B должен быть с севера.Но тогда А — зеленый рассказчик правды, а значит, тоже с севера — противоречие. Итак, А красный. Следовательно, B зеленый и тоже говорит правду. Итак, B — зеленый северянин, а A — красный южанин (поскольку A тоже говорит правду).
Остров мечтателей Решение
(I) Житель должен бодрствовать, чтобы верить в то, что он / она ведет дневной образ жизни, и должен вести ночной образ жизни, чтобы верить в то, что он / она спит.
(II) Хотя мы точно не знаем, во что верила жена, мы знаем, что муж считал их обоих ночными.В частности, он считал, что ведет ночной образ жизни. Значит, он, должно быть, спал, а это значит, что его жена не спала. И хотя мы не можем определить тип мужа и жены, мы можем сделать вывод, что они одного типа. Если бы они были разных типов, то, поскольку один бодрствовал, а другой спал, они бы согласились в своих убеждениях (то есть противоположные типы в противоположных условиях будут соглашаться, в то время как одни и те же типы в противоположных условиях не будут соглашаться).
Варианты.
Вопрос здравомыслия
В конкретном психиатрическом лечебнице каждый человек принадлежит к одному из двух типов — врач или пациент. Каждый человек также либо полностью вменяем (абсолютно точен в своих убеждениях), либо полностью безумен (совершенно неточен в своих убеждениях — то есть каждое истинное суждение считается ложным, а каждое ложное суждение считается истинным) . Все абсолютно честны — все говорят то, во что искренне верят.
Вы встречаетесь в этом приюте с двумя людьми.Мистер Джонс говорит вам, что мистер Смит — врач в штате. Мистер Смит говорит вам, что мистер Джонс — пациент. Вы обеспокоены тем, что в этом приюте есть проблема — либо там работает сумасшедший врач, либо в него поступил совершенно нормальный пациент. Докажите, что ваша озабоченность обоснована.
Ответ: Предположим, Смит действительно врач. Тогда мистер Джонс в здравом уме. Если доктор Смит также вменяемый, то он правильно говорит вам, что Смит (который в здравом уме) также является пациентом. Другая возможность заключается в том, что Dr.Смит безумен.
Теперь предположим, что Смит не врач. Тогда мистер Джонс сошел с ума. Если мистер Джонс — врач, тогда у нас в штате есть сумасшедший доктор. Если мистер Джонс не врач, то то, что вам сказал мистер Смит, правда, поэтому он вменяемый пациент. Во всех случаях мы приходим к тревожному выводу.
(I) Позже вы встречаетесь с двумя людьми, A и B, и определяете следующее:
A: считает, что B безумен B: считает, что A — врач.
Одному из этих лиц не место в приюте. Кого и почему?
(II) Сделайте заявление о том, что, если вы слышите это, вы знаете, что говорящий — вменяемый пациент (и, следовательно, его следует отпустить).
Решения представлены ниже.
Остров вопрошающих
На этом острове жители не отвечают на вопросы, а задают их. На самом деле они общаются, только задавая вопросы. Неудивительно, что есть два типа жителей — тип «Y» и тип «N».Жители типа «Y» задают только те вопросы, на которые можно правильно ответить «да», в то время как жители типа «N» задают только вопросы, на которые можно правильно ответить «нет».
Житель вас спрашивает: «Я типа Y?» Какие выводы можно сделать?
Рассказываю вам время, когда один из жителей задал мне следующий вопрос: «Я типа N?» Какие выводы можно сделать?
По первой ситуации мы ничего не можем сделать. Житель типа Y может задать этот вопрос, потому что правильный ответ — «да», в то время как житель типа N также может задать этот вопрос, потому что правильный ответ — «нет».Во второй ситуации можно сделать вывод, что я лгу. Такое заявление не может произнести обыватель, независимо от его типа. Если он произнесен типом Y, правильный ответ — «нет», а если произнесен типом N, то правильный ответ — «да».
(I) Вайолет и Итан — жители острова. Вы слышите, как Итан спрашивает: «Мы с Вайолет оба типа N?» Можете ли вы определить их типы?
(II) Артур и Роберт — братья на острове. Вы слышите, как Артур спрашивает Роберта: «По крайней мере, один из нас относится к типу N?» Какие выводы можно сделать?
(III) Муж спрашивает жену: «Милая, мы разные?» Какие выводы можно сделать?
Снова решения представлены ниже.
Левши и правши
А теперь представьте остров, где все жители либо правши, либо левши (никто не владеет обеими руками). Конечно, от «острова Смуллян» мы ожидаем логического поворота, и этот остров ничем не отличается. Здесь все, что кто-то пишет более сильной рукой, верно, а утверждения, написанные более слабой рукой, ложны.
Вы нашли клочок бумаги с заявлением. Вы можете сделать вывод, что писатель левша.Приведите пример утверждения, которое выдает волю автора.
Ответ: «Я написал это левой рукой».
Правша не могла написать такое заявление. Если написано левой рукой, это будет правда, но должно быть ложью, а если написано правой рукой, это будет ложью, но должна быть правдой. С другой стороны (каламбур), левша может написать такое утверждение любой рукой.
(I) Приведите пример предложения, которое должно быть написано правшой, использующей левую руку.(См. Решение на веб-сайте.)
Предположим, вас вызывает полиция для помощи в расследовании уголовного дела. Вам вручают два клочка бумаги с заявлениями, написанными подозреваемым в преступлении. В записках написано:
«Я всегда пишу правой рукой». «Иногда пишу левой рукой».
Полиции необходимо знать, пишет ли подозреваемый левой рукой. Он?
Ответ: Да. Поскольку два утверждения противоречат друг другу, одно должно быть ложью, а другое — правдой.В любом случае оба утверждения не могут быть написаны одной рукой. Таким образом, подозреваемый действительно пишет обеими руками.
(II) В другом случае, когда преступник был известен как левша, полиция обнаружила записную книжку у подозреваемого. Все стороны согласились, что подозреваемый был автором записной книжки. На странице 1 было утверждение: «Предложение на странице 2 ложно». На странице 2 было высказывание: «Предложение на странице 1 написано моей левой рукой». Следует ли полиции задержать подозреваемого?
Решения, представленные ниже.
Решения вариаций
Решение вопроса здравомыслия
(I) Предположим, А. вменяемый. Тогда B безумен и, следовательно, ошибается, полагая, что A — врач. Значит, А не принадлежит, будучи вменяемым пациентом. Теперь предположим, что А. сумасшедший. Тогда B должен быть здравомыслящим и правым, полагая, что A — врач — сумасшедший доктор, которому не место в приюте.
(II) Вот утверждение, которое работает: «Либо я сошел с ума, либо я пациент». Здравомыслящий врач не произнесет это неверное утверждение, в то время как сумасшедший врач не произнесет этого правильного утверждения.Точно так же сумасшедший пациент не произнесет это правильное утверждение, поэтому единственная возможность, которая не приводит к противоречию, — это то, что утверждение произнесено здоровым пациентом.
Остров вопросов, решение
(I) Если Итан принадлежит к типу Y, то ответ на его вопрос должен быть «да», но правильный ответ — «нет». Итак, Итан относится к типу N, ответ должен быть «нет», и поэтому Вайолет относится к типу Y.
(II) Если Артур принадлежит к типу N, то требуемый ответ «нет» приводит к противоречию.Итак, Артур должен принадлежать к типу Y, правильный ответ должен быть «да», что означает, что Роберт относится к типу N.
(III) Хотя мы не можем определить тип мужа, мы можем заключить, что жена принадлежит к типу N. Если муж относится к типу Y, то ответ «да» означает, что они принадлежат к разным типам. Если муж относится к типу N, ответ «нет» означает, что они того же типа.
Решение для левшей / правых
(I) Вот фраза, которая работает: «Я левша, и я написал это предложение правой рукой.«Левша не может написать это предложение ни одной рукой — левой рукой оно будет ложным, а правой — истинным. Таким образом, предложение должно быть написано правшой. Поскольку предложение , написано правшой, является ложным (независимо от того, какой рукой он написан), он должен быть написан левой рукой.
(II) Подозреваемый правша и должен быть освобожден. Мы можем определить это, даже если мы не всегда можем определить, истинны или ложны указанные утверждения.Если предложение на странице 1 верно, то предложение на второй странице ложно, что означает, что предложение на странице 1 было написано правой рукой подозреваемого. Итак, подозреваемый написал правдивое заявление на странице 1 правой рукой, а значит, и правшой. В качестве альтернативы, если утверждение на странице 1 неверно, то предложение на странице 2 верно. Таким образом, подозреваемый использовал свою левую руку, чтобы написать ложное заявление на странице 1, снова сделав подозреваемого правшой.
Пазлы Knights and Knaves — puzzlewocky
Эти головоломки связаны с странным островом, населенным двумя типами людей: людьми, которые говорят только правду (рыцари) и людьми, которые лгут только (лжецы).Раймонд Смуллян собрал десятки подобных головоломок в своей книге «Как называется эта книга?». Следующий список быстро меняется от простого к очень сложному. Когда вы разберетесь с ними, попробуйте самую сложную логическую головоломку на свете.
1. Перед вами стоят два человека, Красный и Синий. Красный говорит: «Мы оба лжецы». Что они на самом деле?
См. Ответ
Красный не может быть рыцарем, потому что тогда он солгал бы, говоря, что он лжец. Следовательно, он лжец, и его заявление — ложь, что означает, что Блю должен быть рыцарем.
2. Снова два человека. Красный говорит: «Мы люди одного типа», а Синий говорит: «Все мы разные люди». Каковы их личности?
См. Ответ
. Поскольку их утверждения противоречат друг другу, должно быть, что один рыцарь, а другой лжец. Следовательно, Синий — рыцарь.
3. Вы подошли к развилке дорог и хотите узнать, какой путь ведет к месту назначения. Там стоят два человека. Вы знаете, что один рыцарь, а другой лжец, но вы не знаете, что есть что.Какой единственный вопрос типа «да» или «нет» вы могли бы задать одному из островитян, чтобы узнать правильный путь?
См. Ответ
. Укажите на путь и спросите одного: «А другой скажет мне, что это правильный путь?» Если ответ отрицательный, это правильный путь; если да, то это неверный путь.
4. Теперь вы встречаетесь с развилкой дороги, но там стоит только один житель, и вы не знаете, рыцарь он или лжец. Какой один вопрос типа «да» или «нет» вы могли бы задать, чтобы узнать правильный путь?
Посмотрите «Точка ответа» на путь и спросите: «Если бы я спросил вас, правильный ли это путь, вы бы ответили« да »?» Вы можете рассматривать ответ «Да» как указание на правильный путь, а «Нет» — как на неправильный путь.Вот почему:
Прежде всего, предположим, что человек — рыцарь и путь правильный. Рыцарь отвечает утвердительно. Если человек рыцарь, а путь неверен, рыцарь ответит «нет». Если человек лжец и путь верен, то лжец солгал бы и сказал «нет», если бы его спросили. Однако вы спрашиваете, ответил бы он «да», если бы его спросили, поэтому он должен солгать и сказать «да». Если человек лжец и путь неверен, то лжец солгал и сказал бы «да», если бы его спросили. Но вы спрашиваете, ответил бы он «да», если бы его спросили, поэтому он должен солгать и сказать «нет».
5. Чтобы усложнить ситуацию, обнаружен новый остров с третьим типом жителей: шпионы, которые могут либо сказать правду, либо солгать. Вы встречаетесь с тремя людьми, по одному от каждого, но не знаете, кто из них какой. Красный говорит: «Я рыцарь». Синий говорит: «Красный говорит правду». Грин говорит: «Я шпион». Кто что?
См. Ответ
Может ли Блю лгать? Это будет означать, что Красный тоже лжет, поэтому Грин должен быть рыцарем. Но Грин не может быть рыцарем, потому что рыцарь не станет лгать о том, кто он такой.Следовательно, Синий говорит правду, поэтому Красный — рыцарь. Поскольку Блю — не рыцарь, который сказал правду, он должен быть шпионом, а Грина — лжецом.
6. Теперь добавим морщинки языковых трудностей. На еще одном острове все жители либо рыцари, либо лжецы, и хотя они понимают английский, они не произносят слов «Да» и «Нет». Они используют «Джа» и «Да», но мы не знаем, что означает «Да» и какой означает Нет. Столкнувшись с жителем этого острова, вы спрашиваете: «Джа означает« да »?» Он отвечает: «Джа.Вы узнали, что имеет в виду Джа? Вы узнали, рыцарь это человек или лжец?
См. Ответ
. Вы не узнали значения Джа и Да, но знаете, что это рыцарь. Если Джа означает «да», то рыцарь ответит Джа (да). Если Джа не означает «да», то рыцарь ответит Джа (нет). Если бы этот человек был лжецом, а Джа имел в виду «да», он бы солгал и сказал Да (нет). Если бы Джа не имел в виду «да», то он солгал бы и сказал «да» (да).
7. Вы встречаетесь с другим жителем того же острова и полны решимости узнать значения Джа и Да.Какой один вопрос типа «да или нет» вы можете задать, чтобы узнать это?
См. Ответ
. Просто спросите: «Вы рыцарь?» Независимо от того, рыцарь ли житель или лжец, он ответит словом «да».
8. Предположим, что вы не определили значения Ja и Da, но встретили одного жителя того же острова на развилке дороги и должны найти правильный путь. Сможете ли вы сделать это с помощью одного вопроса «да» или «нет»?
См. Ответ Вы можете заставить жителя указать правильный путь, даже не зная личности человека или значения Джа и Да.Укажите на путь и спросите: «Если бы я спросил вас, правильный ли это путь, вы бы сказали Джа?» Если ответ — Джа, это правильный путь. Если ответ — Да, это неправильный путь. Вот почему:
Если человек рыцарь и это правильный путь, и Джа означает «Да», то он ответит Джа. Если человек рыцарь и это правильный путь, а Джа означает «Нет», то он ответит Джа (Нет, я бы не ответил «нет»).
Если человек рыцарь и это неправильный путь, и Джа означает «да», то он ответит Да (нет).Если человек рыцарь, и это неправильный путь, а Джа означает «нет», то он ответит «Да» (да, я бы сказал «нет»).
Если человек лжец и это правильный путь, а Джа означает «да», то он солгает и ответит Джа (да, я бы сказал «да»). Если человек лжец, и это правильный путь, а Джа означает «нет», то он солгает и ответит Джа (нет, я бы не сказал «нет»).
Если человек лжец и это неправильный путь, а Джа означает «да», то он солгает и ответит Да (нет, я бы не сказал «да»).Если человек лжец, и это неправильный путь, а Джа означает «нет», то он солгает и ответит Да (да, я бы сказал «нет»).
Knights and Knaves — newheiser
Ни B, ни C не могут быть рыцарем. C не может, потому что он утверждает, что является шпионом, а B не может, потому что он утверждает, что кто-то другой является рыцарем. Следовательно, A — рыцарь. B говорит правду, значит, он, должно быть, шпион, а C — лжец.
В этом случае B не может быть шпионом, потому что тогда и рыцарь, и лжец обвинят его в шпионаже.И если B не шпион, то ни A, ни C не могут быть рыцарями, поскольку они не будут говорить правду. Следовательно, B — рыцарь, поэтому C — шпион, а A — лжец.
Есть только шесть возможных комбинаций рыцаря, лжеца и шпиона, которые вы можете использовать для конкретной головоломки:
Любое утверждение, сделанное кем-либо в головоломке, может быть классифицировано в зависимости от того, какие возможности оно исключает. Например, утверждение «Я шпион» оставляет следующие возможности, поскольку заявление может быть сделано только мошенником или шпионом:
Шпион может сделать любое заявление, ( при условии, что шпионы не подлежат такие парадоксы, как «это утверждение ложно» или «утверждение, что я рыцарь, будет иметь такую же ценность истинности, как и это утверждение».Возможны 4 или 16 различных уникальных утверждений, в зависимости от того, какую из 4 перестановок ролей это утверждение исключает.
В следующем списке договоренности представлены в следующей форме: TFS означает «Истинный ложный шпион», то есть A будет рыцарем, B — лжецом, а C — шпионом. Для каждого из следующих действий порядок основан на предположении, что A делает заявление.
Утверждения помещаются рядом с их дополнениями (например, «Я рыцарь» и «Я не рыцарь» помещаются вместе, поскольку каждое утверждение имеет дополнительные наборы возможностей).Следовательно, если вы зададите вопрос формы «Вы рыцарь?» вам гарантирован один из двух ответов и результирующий набор возможностей.
По возможности утверждения также располагаются рядом с эквивалентной формой, в которой вопрос задается о B, а не о C, например, «B — рыцарь» против «C — рыцарь», в которой возможности для B и C равны просто перевернул.
Для всего нижеприведенного предполагается, что А является говорящим, утверждения можно переназначить другому говорящему, изменив ссылки на B / C соответствующим образом и переставив значения в наборе возможностей.
Бесполезное заявление: Я рыцарь
«Я рыцарь» или «Я не лжец». Любая роль может сделать это заявление, поэтому это исключает возможности договоренностей рыцарей, лжецов и шпионов.
Аналогично: «Я лжец». Это утверждение означает, что А должен быть шпионом, поскольку ни рыцарь, ни лжец не могут сделать это заявление.
Возможные варианты: SFT, STF
«Истинное» утверждение: я не шпион
В эту категорию включается любое утверждение, которое не может произнести мошенник, например, вода мокрая и т. Д.
Возможные варианты: TFS, TSF, SFT, STF
«Ложное» заявление: я шпион
Это утверждение, которое не мог бы сказать рыцарь, например, вода не мокрая и т. Д.
Возможные варианты: FTS, FST, SFT, STF
B — лжец
Это утверждение исключает возможность Knight, Spy, Knave.
Возможные варианты: TFS, FTS, FST, SFT, STF
B — не лжец
Это утверждение исключает возможность того, что говорящий лжец, а B — лжец, если говорящий — рыцарь.
Возможные варианты: TSF, SFT, STF
C — лжец
Это утверждение исключает возможность Knight, Knave, Spy
Возможные варианты: TSF, FTS, FST, SFT, STF
C не лжец
Это утверждение означает, что говорящий либо шпион, либо единственная комбинация — это Knight Knave Spy
Возможные варианты: TFS, SFT, STF
B — рыцарь
Это утверждение означает, что говорящий либо шпион, либо единственная комбинация это Валет, Шпион, Рыцарь
Возможные варианты: FST, SFT, STF
B не рыцарь
Это утверждение исключает возможность использования Валет, Шпион, Рыцарь
Возможные варианты: TFS, TSF, FTS, SFT, STF
C — рыцарь
Это утверждение означает, что говорящий либо шпион, либо единственная комбинация — Knave Knight Spy
Возможные варианты: FTS, SFT, STF
C не рыцарь
Это утверждение el воображает возможность Валета, Рыцаря, Шпиона.
Возможные варианты: TFS, TSF, FST, SFT, STF
B — шпион (или C не шпион)
Это утверждение означает, что у вас не может быть Knight Knave Spy или Knave Spy Knight.
Возможные варианты: TSF, FTS, SFT, STF
B не шпион (или C — шпион)
Это утверждение означает, что у вас не может быть Knight Spy Knave или Knave Knight Spy
Возможные варианты: TFS , FST, SFT, STF
Краткое содержание
Приведенные выше утверждения охватывают все возможные схемы, кроме двух, где у вас остались TSF FST или TFS FTS.Оба эти утверждения гарантируют, что кто-то не является шпионом, независимо от того, кто их говорит.
Последние два утверждения обладают тем свойством, что рыцари / лжецы дают идентичный ответ на поставленный вопрос, а утверждение раскрывает что-то о другой роли, не раскрывая ничего о говорящем.
Если вы спросите меня, я бы сказал, что B — шпион (или «если бы вы спросили, я бы сказал, что C не шпион»)
Это утверждение означает, что C не является шпионом.
Возможные договоренности: TSF, FST, SFT, STF
Если вы спросите меня, я бы сказал, что B не шпион (или «если вы спросите, я бы сказал, что C — шпион»)
Это утверждение означает, что Б не шпион.
Возможные договоренности: TFS, FTS, SFT, STF
Если шпион говорит первое утверждение, C не может быть шпионом, поскольку A — шпион. Если рыцарь говорит первое утверждение, C не может быть шпионом, поскольку B должен быть шпионом. И если мошенник говорит это заявление, это означает, что он говорит вам, что он сказал бы, если бы вы спросили, что означает, что он лжет о лжи, которую он скажет, что означает, что он говорит правду, поэтому в этом случае B будет шпионом.
По сути, он использует двойное отрицание, чтобы обойти тот факт, что лжец обычно лжет.«Если и только если это утверждение ложно, B — шпион» также будет работать, но в основном нет реального способа выполнить одно из этих утверждений без ссылки на себя или утверждения типа «B сказал бы».
Интересно отметить, что утверждение B как шпиона эквивалентно утверждению C не с точки зрения его последствий, независимо от того, заявлено ли это нормально или в форме «если вы спросили». Это интересно, потому что, хотя эти два понятия логически эквивалентны с точки зрения человека, приближающегося к проблеме, такое поведение не гарантируется: «Я шпион» не эквивалентно утверждению «Б не шпион», Например.Просто рыцари и лжецы гарантированно сделают противоположные утверждения о двух других людях относительно того, являются ли они шпионами.
Хорошая вещь во всей этой настройке состоит в том, что, охватывая последние два случая, любой оператор можно свести к одной из вышеуказанных форм на основе перестановок, которые он исключает.
Возможности
Поскольку у нас есть 16 возможных утверждений и три человека, которые их решают, у нас есть 4096 возможных головоломок. Многие из них можно уменьшить. Не считая 16 из них, где все говорят одно и то же, у вас есть двусторонняя симметрия в 720 из них и трехсторонняя симметрия в 3360 из них.Это сокращается до 560 + 240+ 16 = 816 уникальных головоломок, если не учитывать перестановки.
После запуска программы для генерации всего набора головоломок и проверки решений из этого общего количества 816 головоломка не имеет единственного решения, а 161 головоломка не имеет решения вообще, что означает парадокс. 274 головоломки имеют уникальное решение и считаются действительными формами головоломки.
Это не может считаться минимальной формой для вариантов головоломки. Бывают случаи, когда A и B говорят одно и то же, в которых C может сделать заявление об A или B, и это сводится к одной и той же загадке.Принимая во внимание симметрию, эти два человека могут сказать 16 вещей, а третье лицо может сказать 12 вещей, которые в конечном итоге эквивалентны. Вычитая 12 вариантов, в которых все они говорят об одном и том же, остается 180 вариантов, половину из которых (90) можно исключить, в результате остается 726 вариантов.
Головоломки без уникального решения
Головоломки без решения
Головоломки с одним уникальным решением (т. Е. Действительные головоломки)
Веселые вопросы
1. Всего за три вопроса определите, кто такой рыцарь, лжец и шпион.Независимо от того, что они ответят, вы сможете выяснить, кто есть кто, задав три вопроса. Вы можете динамически выбирать и направлять свои вопросы.
2. Всего за два вопроса выясните, кто шпион. Ваши вопросы могут принимать любую форму, и их можно выбирать и направлять в зависимости от того, какие ответы вы получите.
3. Для этой и следующей проблемы предположим, что мы работаем с набором вопросов, который не включает два последних утверждения в форме «что бы вы сказали, если бы». При таком ограничении всегда можно решить головоломку всего за три вопроса? Какое минимальное количество вопросов для идентификации шпиона и каждого человека?
4.Вы рыцарь, а ваш друг лжец берет интервью у вас и у шпиона, чтобы узнать, кто есть кто. Шпион делает случайное заявление, например: «Я не шпион». Можете ли вы и этот мошенник с помощью одного заявления ясно заявить о своей личности? Сможете ли вы сделать это при любом возможном заявлении шпиона?
5. Предположим, что вместо того, чтобы иметь дело с тремя людьми, вы должны выяснить, кто такие рыцари, лжецы и шпионы из произвольного числа людей. Одна вещь, которую вы можете заметить по поводу этого типа проблем, заключается в том, что, если у вас есть большинство шпионов, их будет невозможно решить.Например, имея 100 возможных кандидатов и 51 шпион, у вас нет возможности узнать правду о чем-либо. Однако, если вы знаете, что шпионов 49 или меньше, вы можете определить, кто есть кто.
Допустим, вы имеете дело с большим количеством людей, порядка тысячи или около того, и вам нужно идентифицировать всех шпионов из этой группы. Вы знаете, что шпионы составляют меньшинство всех людей. Найдите быстрый и эффективный метод определения того, является ли каждый человек шпионом или нет, не задавая лишних вопросов.
Должно быть не более 5 * (количество человек) вопросов. (линейное время, другими словами)
(Этой последней проблемой занялся математик, доктор Марк Уайлдон из Университета Суонси, которому удалось найти оптимальное решение: http://www-maths.swan.ac.uk/staff/mjw/knights.html)
, напишите мне
Конец заметок
Не так много интересных головоломок, в которых говорят только один или два человека, и технически эти головоломки являются подмножеством головоломок, в которых все три человека говорят в любом случае, так как вы можете просто взять все головоломки, в которых А говорит: «Я рыцарь», и из оставшихся ответов построить головоломку с двумя лицами, поскольку утверждение А не раскрывает никакой информации.
У вас могут быть головоломки, в которых два или более человека делают несколько утверждений, с практическим пределом в 6 утверждений, поскольку, если какое-либо отдельное утверждение не устраняет ни одну из возможностей, его можно просто исключить из головоломки. N утверждений, где N — это количество возможных конфигураций для этого типа головоломки.
Математика | Бесплатный полнотекстовый | Головоломки правды – лжеца с самооценкой
3.4. Анализ SSS-головоломок
В отличие от теоремы 1, если мы рассматриваем головоломку из примера 2 как SSS-головоломку, то эта головоломка имеет ровно одно решение (см. Также пример 4 ниже). Этот факт является не только важным свойством SSS-головоломок, но также подчеркивает различную природу SS- и SSS-головоломок. С другой стороны, теорема 1 имеет прямое следствие для наших SSS-головоломок.
Следствие 1.
В решении хорошей и понятной SSS-головоломки C ≠ {}.
Пример 4.
Пусть дана SSS-головоломка с тремя людьми: Марк сказал, что Антоний — Лжец. Сара сказала, что Марк — лжец, а Антоний сказал: «Я лжец». Кто такой лжец, кто говорит правду, а кто сумасшедший?
У этой загадки есть одно решение: Антоний — Сумасшедший, Марк — Лжец, а Сара — Говорящая Правду. Это потому, что Антоний сказал противоречивое заявление, а Марк сказал ложное заявление об Антонии, поэтому Марк — Лжец, а Сара — Говорящая правду, потому что она сказала истинное атомарное утверждение о Марке.
Поскольку сумасшедшие люди играют важную роль в поиске решений, позвольте нам их идентифицировать. Первый шаг — про Сумасшедших, которые не молчат в головоломке.
Лемма 2.
В разрешимой SSS-головоломке с множеством N людей и их атомарных утверждений, если Ai∈Γi, 0, то Ai∈C.
Доказательство.
Исправим решение. Если (Ai∈Γi, 0) ∈α, то Ai∈T, поскольку он / она сказал истинное атомарное утверждение, но на самом деле Ai сказал о себе / себе, что он / она является лжецом, что означает, что Ai∈L .Отсюда следует, что (Ai∈Γi, 0) ∈β, что противоречит нашему предположению (Ai∈Γi, 0) ∈α. Если мы предположим (Ai∈Γi, 0) ∈β, то Ai∈L, поскольку он / она сказал ложное атомарное утверждение, но, поскольку Ai сказал о себе / себе, что он / она является лжецом, Ai∈T, что означает что (Ai∈Γi, 0) ∈α, что противоречит нашему предположению (Ai∈Γi, 0) ∈β. Следовательно, единственная возможность — (Ai∈Γi, 0) ∈γ, что означает, что Ai∈C. □
По лемме 2 в решении примера 4 Антоний — сумасшедший, так как он сказал утверждение «Я лжец.”
Лемма 3.
Пусть дана разрешимая SSS-головоломка с / решением. Если тип Aj совпадает с типом Ak (Aj, Ak∈N, j ≠ k) в решении, то Aj∉Γk, 0 и Ak∉Γj, 0.
Доказательство.
Исправим решение. Пусть Aj, Ak∈N (j ≠ k) такие, что хотя бы один из Aj и Ak произносит какое-то утверждение. Предположим, что Aj∈Γk, 0 (или Ak∈Γj, 0). Тогда, если Aj, Ak∈C, это означает, что Aj, Ak сказали только самопротиворечивые утверждения, но Aj∈Γk, 0 (или Ak∈Γj, 0) является ложным атомарным утверждением, что означает, что это утверждение не является самопротиворечивым. -противоречие, что противоречит предположению, что Aj, Ak∈C.Если мы предположим, что Aj, Ak∈T, то Aj∈Γk, 0 (или Ak∈Γj, 0) означает, что Aj∈Γk, 0 (или Ak∈Γj, 0) является ложным атомарным утверждением, сказанным Истинным Теллером. это тоже противоречие. Наконец, если Aj, Ak∈L, то из Aj∈Γk, 0 (или Ak∈Γj, 0) следует, что (Aj∈Γk, 0) ∈α (или (Ak∈Γj, 0) ∈α), что означает, что Aj∈Γk, 0 (или Ak∈Γj, 0) — истинное атомарное утверждение, сказанное Лжецом, что противоречит определению 9. Наконец, если ни Aj, ни Ak не говорят ни одного утверждения в головоломке, то, очевидно, оба Aj∉Γk, 0 и Ak∉Γj, 0 выполняются. Следовательно, не существует решения SSS-головоломки, в которой два человека, Aj, Ak, имеют один и тот же тип и Aj∈Γk, 0 (или Ak∈Γj, 0).□
Следовательно, по лемме 3, если головоломка разрешима, то в графическом представлении G нет разорванного ребра между любыми двумя людьми одного типа (т.е. оба они говорят правду, оба лжецы или оба сумасшедшие) в решении головоломки.
Следствие 2.
В SSS-головоломке, если есть два человека Ai и Aj такие, что Ai∈Γj, 0 (или Aj∈Γi, 0), и оба Ai и Aj сказали одно и то же утверждение о от третьего лица Ак, то загадка неразрешима.
Как следует из следствия, существуют SSS-головоломки без решения. Мы уже видели, что некоторые из SS-головоломок, у которых нет решений, становятся разрешимыми, если мы думаем о них как о SSS-головоломках, то есть если мы допускаем не только Истинщиков и Лжецов, но и Сумасшедших людей в цель функции решения. . Оказывается, таким способом можно решить не каждую головоломку.
Мы продолжаем выяснять, кто такие Сумасшедшие в решениях.
Лемма 4.
В решении SSS-головоломки с множеством лиц N для человека Ai∈N, если
Γi, 0 = ∅, Γi, 1 = ∅и
∃ Aj, Ak∈N / {Ai} такое, что тип Aj совпадает с типом Ak, и Ai∈Γj, 0, Ai∈Γk, 1 (или Ai∈Γk, 0 и Ai∈Γj, 1),
, тогда Ai∈CandAj, Ak∈L .
Доказательство.
В случае, если j ≠ k и тип Aj такой же, как тип Ak, но они говорят два разных утверждения об Ai («Ai — лжец» и «Ai — рассказчик правды»), затем, чтобы имеют решение Aj, Ak∈L и Ai∈C; в противном случае загадка неразрешима.
Если j = k, то это означает, что есть один человек Aj, который сказал два разных утверждения об Ai. Следовательно, чтобы иметь решение, Aj∈L и Ai∈C; в противном случае загадка неразрешима. □
Теперь мы готовы дать характеристику Сумасшедших.
Теорема 2.
В решаемой SSS-головоломке с множеством людей N, если Ai∈C, то Ai может сказать не более одного атомарного утверждения, которое является Ai∈Γi, 0.
Доказательство.
В SSS-головоломке любой человек Ai может произнести только две формы элементарных утверждений: либо Aj∈Γi, 0, либо Aj∈Γi, 1, где Aj∈N.Предположим, что Ai∈C и Aj∈Γi, 0, где i ≠ j. Если Aj∈L, то (Aj∈Γi, 0) ∈α, что противоречит Ai∈C. Если Aj∈T, то (Aj∈Γi, 0) ∈β, что противоречит Ai∈C. Если Aj∈C, то (Aj∈Γi, 0) ∈β; таким образом, (Aj∈Γi, 0) ∉γ и Ai∉C.
Предположим теперь, что Ai∈C и Aj∈Γi, 1 с i ≠ j. Если Aj∈L, то (Aj∈Γi, 1) ∈β, что противоречит Ai∈C. Если Aj∈T, то (Aj∈Γi, 1) ∈α; таким образом, Ai∉C. Если Aj∈C, то (Aj∈Γi, 1) ∈β; таким образом, (Aj∈Γi, 1) ∉γ и Ai∉C. Если Ai∈C, то (Ai∈Γi, 1) ∈β, что также противоречит Ai∈C.
Следовательно, единственное утверждение, которое сумасшедший может сказать Ai, — это Ai∈Γi, 0, что является противоречивым утверждением.□
На основании теоремы 2 и леммы 2, если Ai∈C, Ai может сказать только Ai∈Γi, 0; или, основываясь на лемме 4, Ai не говорит никаких атомарных утверждений о других людях.
Для дальнейшего анализа мы используем графические представления головоломок.
Определение 12.
Решение графа G — это функция, которая назначает C, L или T каждому узлу, так что все утверждения, представленные ребрами графа, удовлетворяются.
Предложение 2.
Решения головоломки такие же, как решения графического представления G головоломки.
Доказательство.
В графе G узлы и края G представляют людей и утверждения в головоломке, которые представлены G, соответственно. Поэтому, основываясь на определениях 9 и 12, множество решений головоломки такое же, как и множество решений G. □
Напомним понятие полных графов из теории графов. Kn — это граф из n узлов, каждая пара узлов которого соединена ребром.В частности, K3, треугольный граф, представляет собой граф, содержащий три узла, причем между любыми двумя вершинами есть ребро. В следующем результате мы покажем, как треугольные графы связаны с решаемыми головоломками.
Предложение 3.
Если графическое представление G SSS-головоломки P содержит подграф, основной неориентированный граф которого представляет собой K3 с сломанными ребрами, то P не имеет решения.
Доказательство. Предположим, что графовое представление G головоломки содержит подграф с сломанными ребрами, такой, что основной неориентированный граф этого подграфа равен K3. Рассматривая этот подграф, есть две возможные формы неизоморфных подграфов в G с сломанными ребрами. .На рисунке 1 показаны эти два подграфа. Начнем с графика, показанного на рисунке 1а. В решении головоломки A и D имеют один и тот же тип, поскольку они говорят об одном и том же утверждении о B. Однако, согласно лемме 3, для того, чтобы иметь решение A∉ΓD, 0 и D∉ΓA, 0, должно быть противоречие в формировании головоломки, края которой представлены подграфом, показанным на рисунке 1a. Рассматривая рисунок 1b в решении головоломки, A∉C, B∉C и D∉C, потому что по теореме 2 Сумасшедший не может сказать никаких утверждений о других людях.Предположим, что A∈T; следовательно, D∈L и B∈L, но B∈ΓD, 0, что противоречит лемме 3. С другой стороны, если предположить, что A∈L, то D∈T и B∈T, но B∈ΓD, 0, что также противоречит лемме 3. □
Последнее предложение полезно, если кто-то хочет предоставить неразрешимую головоломку, используя ее графовое представление.
Курс логики BEAM Discovery — Головоломки лжецов и правдивых — Переход к продвинутой математике
Что такое головоломки лжецов и правдивых?
В головоломках лжецов и правдивых вы сталкиваетесь с ситуацией, когда вы разговариваете с людьми, которые либо всегда лгут, либо всегда говорят правду.Их часто представляют как пример, когда вы находитесь на «острове рыцарей и лжецов», где рыцари всегда говорят правду, а лжецы всегда лгут (а нормальные люди могут говорить правду или лгать). Вы должны сделать некоторый вывод на основании утверждений, сделанных разными людьми на острове. Классическая головоломка может быть такой:
Среди трех людей A, B и C один рыцарь, один лжец и один нормальный.
A говорит: «Я нормальный». Тогда B говорит: «Это правда». Затем C говорит: «Я ненормальный.
Что такое A, B и C?
Насколько математически ценны головоломки лжецов и правдивых?
Головоломки учат дедуктивной логике и рассуждениям непосредственно по случаям. Например, в приведенной выше головоломке мы сразу же делаем вывод, что A есть не рыцарь, потому что рыцарь никогда бы не сказал: «Я нормальный». Если бы А был нормальным, то Б не мог бы быть лжецом, поскольку они подтверждают, что А говорит правду. Это сделало бы В рыцарем, а С — Лжец. Для C невозможно быть лжецом, потому что C сказал бы правду, сказав: «Я ненормальный.»Следовательно, A тоже не может быть нормальным, и мы знаем, что A — лжец. Мы уже видели анализ случаев, логический вывод и встроенное доказательство от противоречия. требуют пристального внимания к деталям; их нельзя просто читать в поисках ключевых фраз, и не существует «единственного способа» их решить. Студенты усваивают идею о том, что утверждениям можно придать значение истинности. Наконец, они изучают математическое значение » if «и чем он отличается от обычного английского.
Например, головоломка «Лжец и правдивый» может перейти к решению этой задачи (из конкурса «Математический кенгуру»):
В коробке 50 кубиков красного, белого и синего цветов. Количество белых кирпичей в одиннадцать раз больше, чем синих. Красных меньше, чем белых, но красных больше, чем синих. Сколько есть кирпичей каждого цвета?
Чтение каждого утверждения, понимание того, что делает его истинным, а затем пошаговый анализ с учетом всех случаев имеет важное значение для этой (и многих других) математических задач.Явно проводя это различие, учащиеся могут увидеть, как решать сложные задачи.
Головоломки «Лжец и правдивый» также можно использовать для начала исследования идеи математических доказательств (и ложных доказательств) с анализа аргументов в пользу истинности или ложности каждого утверждения.
Как может работать этот класс?
В конечном счете, класс принадлежит вам, чтобы вести его так, как вы считаете, лучше всего подходит для учащихся. Вот один из возможных графиков.
3 дня: Решите несколько головоломок лжецов и правдивых, создавая более сложные варианты.Хорошим источником является книга «Как называется эта книга?». Раймонда Смулляна.
2 дня: В зависимости от успеваемости ученика, продолжайте разгадывать головоломки лжеца и рассказчика правды или вводите другой тип головоломки.

 Давай-ка разберем в этой статье отрицание в немецком: kein и nicht, узнаем, когда и что нужно употреблять!
Давай-ка разберем в этой статье отрицание в немецком: kein и nicht, узнаем, когда и что нужно употреблять!


 Перемалывание зёрен в муку, падение камня, кипение воды, молния, свечение лампочки, растворение сахара в чае, движение транспортных средств, молнии, радуги – это примеры физических явлений.
Перемалывание зёрен в муку, падение камня, кипение воды, молния, свечение лампочки, растворение сахара в чае, движение транспортных средств, молнии, радуги – это примеры физических явлений. Подобно воде, и другие вещества можно переводить из одного агрегатного состояния в другое.
Подобно воде, и другие вещества можно переводить из одного агрегатного состояния в другое. Магнитные явления – это явления, связанные с возникновением у физических тел магнитных свойств (притяжение магнитом железных предметов, поворот стрелки компаса на север).
Магнитные явления – это явления, связанные с возникновением у физических тел магнитных свойств (притяжение магнитом железных предметов, поворот стрелки компаса на север).
 Для каждого из равенств, например, для основного свойства, можно поменять местами правую и левую часть: am·an=am+n — то же самое, что и am+n=am·an. В таком виде оно часто используется при упрощении выражений.
Для каждого из равенств, например, для основного свойства, можно поменять местами правую и левую часть: am·an=am+n — то же самое, что и am+n=am·an. В таком виде оно часто используется при упрощении выражений.


 Укажем разность и вынесем an за скобки: (am−n−1).Степень an при а, большем единицы, даст положительный результат; а сама разность также окажется положительна в силу изначальных условий, и при a>1 степень am−n больше единицы. Выходит, am−an>0 и am>an, что нам и требовалось доказать.
Укажем разность и вынесем an за скобки: (am−n−1).Степень an при а, большем единицы, даст положительный результат; а сама разность также окажется положительна в силу изначальных условий, и при a>1 степень am−n больше единицы. Выходит, am−an>0 и am>an, что нам и требовалось доказать.
 Их свойства такие же, что и у степеней с целыми показателями. Запишем:
Их свойства такие же, что и у степеней с целыми показателями. Запишем: При этом m–целое число, n–натуральное. Тогда условия p<0 и p>0 будут распространяться на m<0 и m>0. При m>0 и a<b имеем (согласно свойству степени с целым положительным показателем), что должно выполняться неравенство am<bm.
При этом m–целое число, n–натуральное. Тогда условия p<0 и p>0 будут распространяться на m<0 и m>0. При m>0 и a<b имеем (согласно свойству степени с целым положительным показателем), что должно выполняться неравенство am<bm.

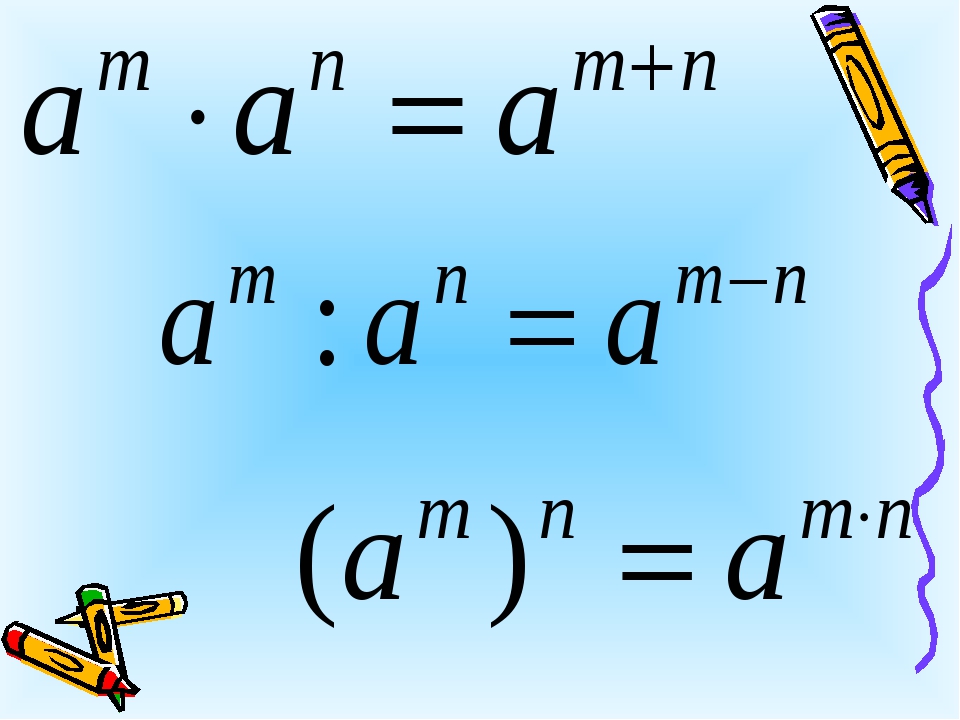









 Критерии оцениванияКоличество
Критерии оцениванияКоличество





 Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода.
Рис. Модель молекулы водорода и её осевая проекция
Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода.
Рис. Модель молекулы водорода и её осевая проекция Молекула дифторида ксенона
Молекула дифторида ксенона  Молекула тетрафторида ксенона
Молекула тетрафторида ксенона  Молекула гексафторида ксенона
Молекула гексафторида ксенона
 Следует отметить, что сатира сама по себе является также разновидностью критики, однако, несколько смягченной по сравнению с ней.
Следует отметить, что сатира сама по себе является также разновидностью критики, однако, несколько смягченной по сравнению с ней. И как любое явление, она имеет свою структуру. В смеховую структуру входит юмор, ирония, остроумие, сатира, сарказм, пародия, карикатура и каламбур. Все данные формы неотъемлемая часть смеховой культуры российского студенчества. Многообразие оттенков смеха (юмор, сатира, ирония, сарказм, шутка, насмешка, гротеск каламбур) отражает богатство действительности. Формы и мера смеха определяются и объективными свойствами предмета, и мировоззренческими принципами человека, его отношением к миру, и национальными традициями культуры народа. Юмор, ирония, сатира, пародия, сарказм предстают как формы целостного смеха. Автор предлагает свою схему структуры смеховой культуры.
И как любое явление, она имеет свою структуру. В смеховую структуру входит юмор, ирония, остроумие, сатира, сарказм, пародия, карикатура и каламбур. Все данные формы неотъемлемая часть смеховой культуры российского студенчества. Многообразие оттенков смеха (юмор, сатира, ирония, сарказм, шутка, насмешка, гротеск каламбур) отражает богатство действительности. Формы и мера смеха определяются и объективными свойствами предмета, и мировоззренческими принципами человека, его отношением к миру, и национальными традициями культуры народа. Юмор, ирония, сатира, пародия, сарказм предстают как формы целостного смеха. Автор предлагает свою схему структуры смеховой культуры. Особенно это проявляется в присутствии других людей, при которых мы стараемся «сохранить» лицо — как говорят японцы. Существуют физиологические, психологические аспекты смеха. С помощью смеха мы освобождаемся от страха, волнения. И в тоже время смех имеет большую социальную значимость. Смех является категорией понимания. Например, юмор студентов может понять лишь студент, смеховая культура того или иного общества несет свои специфические черты. Одновременно с этим, смех можно рассматривать как понимание того, каким образом можно решить проблему, как выйти из сложной и запутанной ситуации.
Особенно это проявляется в присутствии других людей, при которых мы стараемся «сохранить» лицо — как говорят японцы. Существуют физиологические, психологические аспекты смеха. С помощью смеха мы освобождаемся от страха, волнения. И в тоже время смех имеет большую социальную значимость. Смех является категорией понимания. Например, юмор студентов может понять лишь студент, смеховая культура того или иного общества несет свои специфические черты. Одновременно с этим, смех можно рассматривать как понимание того, каким образом можно решить проблему, как выйти из сложной и запутанной ситуации. Юмор — признак самой жизни в ее развитии, движении, переломах и конфликтах, противостоящий всякой статике и теоретизированию.
Юмор — признак самой жизни в ее развитии, движении, переломах и конфликтах, противостоящий всякой статике и теоретизированию. Ирония является выражением объективного контраста, скрывающего за собой субъективность. В иронии смешное скрывается под маской серьёзности — с преобладанием отрицательного (насмешливого) отношения к предмету; в юмор серьёзное — под маской смешного, обычно с преобладанием положительного («смеющегося») отношения. Сложность иронии лишь формальная, её серьёзность — мнимая, её природа — чисто артистическая; напротив, сложность юмора содержательная, его серьёзность — подлинная, его природа — даже в игре — скорее «философическая», мировоззренческая. Юмор нередко «играет» на двух равно действительных аспектах человеческой натуры — физическом и духовном. Различен поэтому эффект иронии и юмора, когда игра закончена и обнажается внутренний аспект, подлинная цель игры: ирония, порой близкая смеху язвительному, задевает, ранит, оскорбляет сугубо — не только открывшимся неприглядным содержанием, но и самой формой игры: тогда как юмор в конечном счёте заступается за свой предмет, а его смехом иногда, например в «дружеском» юмор, «стыдливо» прикрывается восхищение, даже прославление. Колоритом юмора художники нового времени часто пользуются — во избежание ходульности или односторонности — для изображения наиболее благородных героев, а также идеальных натур «простых людей», национально или социально характерных.
Ирония является выражением объективного контраста, скрывающего за собой субъективность. В иронии смешное скрывается под маской серьёзности — с преобладанием отрицательного (насмешливого) отношения к предмету; в юмор серьёзное — под маской смешного, обычно с преобладанием положительного («смеющегося») отношения. Сложность иронии лишь формальная, её серьёзность — мнимая, её природа — чисто артистическая; напротив, сложность юмора содержательная, его серьёзность — подлинная, его природа — даже в игре — скорее «философическая», мировоззренческая. Юмор нередко «играет» на двух равно действительных аспектах человеческой натуры — физическом и духовном. Различен поэтому эффект иронии и юмора, когда игра закончена и обнажается внутренний аспект, подлинная цель игры: ирония, порой близкая смеху язвительному, задевает, ранит, оскорбляет сугубо — не только открывшимся неприглядным содержанием, но и самой формой игры: тогда как юмор в конечном счёте заступается за свой предмет, а его смехом иногда, например в «дружеском» юмор, «стыдливо» прикрывается восхищение, даже прославление. Колоритом юмора художники нового времени часто пользуются — во избежание ходульности или односторонности — для изображения наиболее благородных героев, а также идеальных натур «простых людей», национально или социально характерных. Смех перестает быть смехом — он трансформируется в осмеяние и моральное неодобрение. Сатира (лат. satira, от более раннего satura — сатура, буквально — смесь, всякая всячина), вид комического; беспощадное, уничтожающее переосмысление объекта изображения (и критики), разрешающееся
Смех перестает быть смехом — он трансформируется в осмеяние и моральное неодобрение. Сатира (лат. satira, от более раннего satura — сатура, буквально — смесь, всякая всячина), вид комического; беспощадное, уничтожающее переосмысление объекта изображения (и критики), разрешающееся Но идеал сатирика выражен через «антиидеал», т. е. через вопиюще-смехотворное отсутствие его в предмете обличения.
Но идеал сатирика выражен через «антиидеал», т. е. через вопиюще-смехотворное отсутствие его в предмете обличения. a, буквально — пение наизнанку), в литературе и (реже) в музыкальном и изобразительном искусстве комическое подражание художественному произведению или группе произведений. Обычно пародия строится на нарочитом несоответствии стилистических и тематических планов художественной формы; два классических типа пародии (иногда выделяемые в особые жанры) — бурлеска, низкий предмет, излагаемый высоким стилем и травестия, высокий предмет, излагаемый низким стилем Осмеяние может сосредоточиться как на стиле, так и на тематике — высмеиваются как заштампованные, отставшие от жизни приёмы поэзии, так и пошлые, недостойные поэзии явления действительности; разделить то и другое иногда очень трудно. Пародироваться может поэтика конкретного произведения автора, жанра, целого литературного
a, буквально — пение наизнанку), в литературе и (реже) в музыкальном и изобразительном искусстве комическое подражание художественному произведению или группе произведений. Обычно пародия строится на нарочитом несоответствии стилистических и тематических планов художественной формы; два классических типа пародии (иногда выделяемые в особые жанры) — бурлеска, низкий предмет, излагаемый высоким стилем и травестия, высокий предмет, излагаемый низким стилем Осмеяние может сосредоточиться как на стиле, так и на тематике — высмеиваются как заштампованные, отставшие от жизни приёмы поэзии, так и пошлые, недостойные поэзии явления действительности; разделить то и другое иногда очень трудно. Пародироваться может поэтика конкретного произведения автора, жанра, целого литературного Карикатура в студенческой среде всегда носила позитивный характер. В широком смысле слова под карикатурой понимают всякое изображение, где сознательно создаётся комический эффект, соединяются реальное и фантастическое, преувеличиваются и заостряются характерные черты фигуры, лица, костюма, манеры поведения людей, изменяются соотношения их с окружающей средой, используются неожиданные сопоставления и уподобления. Карикатура в этом значении обладает широчайшим диапазоном тем и может быть сопоставлена с карнавальным действом, театральной буффонадой, литературным бурлеском и эпиграммой.
Карикатура в студенческой среде всегда носила позитивный характер. В широком смысле слова под карикатурой понимают всякое изображение, где сознательно создаётся комический эффект, соединяются реальное и фантастическое, преувеличиваются и заостряются характерные черты фигуры, лица, костюма, манеры поведения людей, изменяются соотношения их с окружающей средой, используются неожиданные сопоставления и уподобления. Карикатура в этом значении обладает широчайшим диапазоном тем и может быть сопоставлена с карнавальным действом, театральной буффонадой, литературным бурлеском и эпиграммой. Эта беспощадная насмешка над тем, чему люди привыкли поклоняться, — дитя общественных разочарований. Утраченные иллюзии стали обыкновенной историей, а в сфере юмора это выразилось в безрадостном, подернутом цинизмом смехе, для которого нет ничего заповедного, неприкосновенного. Характерный пример благга: «Эта женщина как республика, она была прекрасна во времена империи».
Эта беспощадная насмешка над тем, чему люди привыкли поклоняться, — дитя общественных разочарований. Утраченные иллюзии стали обыкновенной историей, а в сфере юмора это выразилось в безрадостном, подернутом цинизмом смехе, для которого нет ничего заповедного, неприкосновенного. Характерный пример благга: «Эта женщина как республика, она была прекрасна во времена империи».
 М. Кубанева»
М. Кубанева»
 Появлялись они только после моих грубых выкриков и громкой брани. Потому что я, оказывается, отнимал у жены последнее. Потому что я, по её мнению, всегда был транжира и мот, который принесет денег с Гулькин нос и тут же их забирает обратно.
Появлялись они только после моих грубых выкриков и громкой брани. Потому что я, оказывается, отнимал у жены последнее. Потому что я, по её мнению, всегда был транжира и мот, который принесет денег с Гулькин нос и тут же их забирает обратно.
 Сегодня я думаю так, завтра – иначе, а через неделю, вполне возможно, заброшу все эти мысли к чертовой бабушке. Я человек непостоянный. Я свободен от каких бы то ни было принципов. А полная свобода, на мой взгляд, как раз и предполагает идейное непостоянство. Сегодня я могу думать так, завтра иначе, а послезавтра вообще могу забыть обо всем, что тревожило меня ранее. Потому что от дум моих ничего не меняется. Думы мои легки и оттого горьки. Из под моей руки – буковки, завитки.
Сегодня я думаю так, завтра – иначе, а через неделю, вполне возможно, заброшу все эти мысли к чертовой бабушке. Я человек непостоянный. Я свободен от каких бы то ни было принципов. А полная свобода, на мой взгляд, как раз и предполагает идейное непостоянство. Сегодня я могу думать так, завтра иначе, а послезавтра вообще могу забыть обо всем, что тревожило меня ранее. Потому что от дум моих ничего не меняется. Думы мои легки и оттого горьки. Из под моей руки – буковки, завитки.
 Он был среди граждан совершенно как родной в семье, а в лавки и в гостиный двор наведывался, как в собственную кладовую.
Он был среди граждан совершенно как родной в семье, а в лавки и в гостиный двор наведывался, как в собственную кладовую.
 В отличие от иронии и сатиры, сарказм применяется в комедиях довольно редко, так как имеет откровенный негативный подтекст.
В отличие от иронии и сатиры, сарказм применяется в комедиях довольно редко, так как имеет откровенный негативный подтекст.



 В ход идут различные стилистические, грамматические приемы, направленные на придание слову, тексту дополнительной выразительности. Одним из таких особых приемов является
В ход идут различные стилистические, грамматические приемы, направленные на придание слову, тексту дополнительной выразительности. Одним из таких особых приемов является При этом в тексте поясняющие слова (как будто, наподобие, напоминающие) отсутствуют, но всегда подразумеваются.
При этом в тексте поясняющие слова (как будто, наподобие, напоминающие) отсутствуют, но всегда подразумеваются.
 Из античности к нам пришло знаменитое утверждение о том, что человек отличается от животного способностью говорить и смеяться. Ф. Рабле называл смех сущностью человека. Религиозный философ В. Соловьев утверждал, что «смех предусматривает свободное состояние и волеизъявление: раб не должен смеяться». По мнению И.В. Гёте, нигде так не проявляется человеческий характер, как в его понимание и находки смешного.
Из античности к нам пришло знаменитое утверждение о том, что человек отличается от животного способностью говорить и смеяться. Ф. Рабле называл смех сущностью человека. Религиозный философ В. Соловьев утверждал, что «смех предусматривает свободное состояние и волеизъявление: раб не должен смеяться». По мнению И.В. Гёте, нигде так не проявляется человеческий характер, как в его понимание и находки смешного. В юморе за смешным нередко прячется грустное, а вот смех саркастический имеет один эмоциональный оттенок — едко-обличительный. Сатире также присуща бескомпромиссность суждений о предмете осмеяния.
В юморе за смешным нередко прячется грустное, а вот смех саркастический имеет один эмоциональный оттенок — едко-обличительный. Сатире также присуща бескомпромиссность суждений о предмете осмеяния. press.uillinois.edu) считается одной из самых крупных и выдающихся университетских издательств страны. Press публикует более 120 новых книг и 30 научных журналов каждый год по множеству предметов, включая историю Америки, историю труда, историю спорта, фольклор, еду, фильмы, американскую музыку, американскую религию, афроамериканские исследования, женские исследования и Авраама. Линкольн.The Press является одним из основателей Ассоциации прессов американских университетов, а также History Cooperative, онлайновой коллекции из более чем 20 журналов по истории.
press.uillinois.edu) считается одной из самых крупных и выдающихся университетских издательств страны. Press публикует более 120 новых книг и 30 научных журналов каждый год по множеству предметов, включая историю Америки, историю труда, историю спорта, фольклор, еду, фильмы, американскую музыку, американскую религию, афроамериканские исследования, женские исследования и Авраама. Линкольн.The Press является одним из основателей Ассоциации прессов американских университетов, а также History Cooperative, онлайновой коллекции из более чем 20 журналов по истории. Другими словами, можно сказать, что сатира Гората развлекается безумием, а сатира Ювеналии атакует преступления или, по крайней мере, преступления, которые считаются антисоциальными.Очевидно, что последний тип, если он вообще вызывает смех, вызывает презрительный смех. Оба типа сатиры встречаются в Candide. И важно то, что даже когда Вольтер был наиболее возбужден, он использовал легкое прикосновение и достигал тона, зачастую веселого, что обманчиво для буквально мыслящего читателя, который принимает сказку как преувеличенное повествование о приключениях главного героя и не более того. . Первичный прием Вольтера как сатирика — это ирония, применяющая ее не только к высказыванию, но и к событию, ситуации и структуре.
Другими словами, можно сказать, что сатира Гората развлекается безумием, а сатира Ювеналии атакует преступления или, по крайней мере, преступления, которые считаются антисоциальными.Очевидно, что последний тип, если он вообще вызывает смех, вызывает презрительный смех. Оба типа сатиры встречаются в Candide. И важно то, что даже когда Вольтер был наиболее возбужден, он использовал легкое прикосновение и достигал тона, зачастую веселого, что обманчиво для буквально мыслящего читателя, который принимает сказку как преувеличенное повествование о приключениях главного героя и не более того. . Первичный прием Вольтера как сатирика — это ирония, применяющая ее не только к высказыванию, но и к событию, ситуации и структуре. Для своей цели Вольтер особенно полагался на преувеличение, но он также использовал контрастирующий прием преуменьшения, часто в форме литотес, который является преуменьшением, при котором что-то утверждается путем констатации отрицания своей противоположности — распространенный прием в ироническом выражении. С ним связан эвфемизм, фигура речи, в которой косвенное утверждение заменяется прямым. Многие авторы использовали эвфемистические термины, чтобы избежать резкости или оскорбления, но они демонстрируют тенденцию к неискренности и сентиментальности.Вольтер использовал их иронично с прекрасным комическим эффектом, чтобы выразить свою сатиру на несправедливость, преступление и глупость. Также следует отметить карикатуру и пародию — способы, с помощью которых автор преувеличивал те или иные детали с той же целью.
Для своей цели Вольтер особенно полагался на преувеличение, но он также использовал контрастирующий прием преуменьшения, часто в форме литотес, который является преуменьшением, при котором что-то утверждается путем констатации отрицания своей противоположности — распространенный прием в ироническом выражении. С ним связан эвфемизм, фигура речи, в которой косвенное утверждение заменяется прямым. Многие авторы использовали эвфемистические термины, чтобы избежать резкости или оскорбления, но они демонстрируют тенденцию к неискренности и сентиментальности.Вольтер использовал их иронично с прекрасным комическим эффектом, чтобы выразить свою сатиру на несправедливость, преступление и глупость. Также следует отметить карикатуру и пародию — способы, с помощью которых автор преувеличивал те или иные детали с той же целью. Мы узнаем, что этот самый красивый и приятный из всех возможных замков, как Вольтер называет его в последнем предложении главы, достаточно груб, что касается его одной двери, окна и одного гобелена.Баронесса страдает ожирением; барон явно примитивный персонаж. Но все эти преувеличения, все превосходные степени готовят читателя к ужасным событиям, которые должны последовать за этим. Точно так же в описании этой неизбывной земли, Эльдорадо, и в описании лесной резиденции дона Иссахара с ее просторными садами и великолепными убранствами Вольтер снова использовал преувеличение как прелюдию к неудачам.
Мы узнаем, что этот самый красивый и приятный из всех возможных замков, как Вольтер называет его в последнем предложении главы, достаточно груб, что касается его одной двери, окна и одного гобелена.Баронесса страдает ожирением; барон явно примитивный персонаж. Но все эти преувеличения, все превосходные степени готовят читателя к ужасным событиям, которые должны последовать за этим. Точно так же в описании этой неизбывной земли, Эльдорадо, и в описании лесной резиденции дона Иссахара с ее просторными садами и великолепными убранствами Вольтер снова использовал преувеличение как прелюдию к неудачам. Часто именно такими лаконичными высказываниями автор добивается остроумной преуменьшенности.
Часто именно такими лаконичными высказываниями автор добивается остроумной преуменьшенности.
 Губернатор Буэнос-Айреса со своими многочисленными именами и невыносимой гордостью стал еще одним примером. Весь обескураживающий эффект в главе I с ее контрастом наивности и догматизма — чистая пародия, особенно ложная трагедия изгнания Кандида из замка.
Губернатор Буэнос-Айреса со своими многочисленными именами и невыносимой гордостью стал еще одним примером. Весь обескураживающий эффект в главе I с ее контрастом наивности и догматизма — чистая пародия, особенно ложная трагедия изгнания Кандида из замка. благородства позади нее! » Позже, когда Кандид и барон, которого Кандид на самом деле не убивал, встретились снова, барон сказал: «Ты можешь убить меня снова, но ты никогда не женишься на моей сестре, пока я еще жив».»Чтобы полностью оценить экстравагантность его слов, нужно вспомнить все, что случилось как с бароном, так и с его сестрой — одна теперь женщина, красота которой полностью поблекла в результате ее страданий, а другая только что спасла Кандид из камбузы.
благородства позади нее! » Позже, когда Кандид и барон, которого Кандид на самом деле не убивал, встретились снова, барон сказал: «Ты можешь убить меня снова, но ты никогда не женишься на моей сестре, пока я еще жив».»Чтобы полностью оценить экстравагантность его слов, нужно вспомнить все, что случилось как с бароном, так и с его сестрой — одна теперь женщина, красота которой полностью поблекла в результате ее страданий, а другая только что спасла Кандид из камбузы. Вот несколько распространенных и знакомых примеров сатиры:
Вот несколько распространенных и знакомых примеров сатиры: Они обращаются к аудитории своим сочетанием внимательности, юмора и критики политики, популярной культуры, социальных условностей, человеческой природы, средств массовой информации и даже самого телевидения. Вот несколько примеров сатирических телевизионных программ:
Они обращаются к аудитории своим сочетанием внимательности, юмора и критики политики, популярной культуры, социальных условностей, человеческой природы, средств массовой информации и даже самого телевидения. Вот несколько примеров сатирических телевизионных программ: Однако между ними есть различия, особенно в их намерениях. Сатира направлена на высмеивание человеческих и / или социальных недостатков, несоответствий и несоответствий как средство провокации аудитории и оспаривания точек зрения. Пародия намеревается имитировать что-то знакомое аудитории в качестве средства развлечения или юмора.
Однако между ними есть различия, особенно в их намерениях. Сатира направлена на высмеивание человеческих и / или социальных недостатков, несоответствий и несоответствий как средство провокации аудитории и оспаривания точек зрения. Пародия намеревается имитировать что-то знакомое аудитории в качестве средства развлечения или юмора. Сатирическая литература привлекает внимание к этим вопросам и может рассказать читателям о том, чего они раньше не рассматривали или не понимали. Это осознание может затем вызвать у читателя призыв к действию, чтобы он осудил, попытался исправить или даже более критически подумать о социальных недостатках.
Сатирическая литература привлекает внимание к этим вопросам и может рассказать читателям о том, чего они раньше не рассматривали или не понимали. Это осознание может затем вызвать у читателя призыв к действию, чтобы он осудил, попытался исправить или даже более критически подумать о социальных недостатках.
 Это позволяет читателю оценить художественный характер любовного стихотворения, одновременно придя к пониманию того, что концепция романтической любви не является устойчивой и является ложной реальностью.
Это позволяет читателю оценить художественный характер любовного стихотворения, одновременно придя к пониманию того, что концепция романтической любви не является устойчивой и является ложной реальностью. Это может показаться сатирой, но это не значит, что это так.
Это может показаться сатирой, но это не значит, что это так. Можно утверждать, что три наиболее распространенных типа сатиры (хоратовская, ювеналийская и мениппийская) теперь смешались и перекрестно опыляются до такой степени, что нередко современное сатирическое произведение представляет собой своего рода гибридную дворнягу.
Можно утверждать, что три наиболее распространенных типа сатиры (хоратовская, ювеналийская и мениппийская) теперь смешались и перекрестно опыляются до такой степени, что нередко современное сатирическое произведение представляет собой своего рода гибридную дворнягу.
 Например, «Алиса в стране чудес» Льюиса Кэрролла — это менипповая сатира в том смысле, что именно любопытство Алисы в конечном итоге приводит к ее тяжелому положению.
Например, «Алиса в стране чудес» Льюиса Кэрролла — это менипповая сатира в том смысле, что именно любопытство Алисы в конечном итоге приводит к ее тяжелому положению.
 Помимо того, что это отдельный жанр, это литературный прием, который часто используется для критики политики и злободневных вопросов.
Помимо того, что это отдельный жанр, это литературный прием, который часто используется для критики политики и злободневных вопросов.
 Мениппейский стиль часто требует большей продолжительности работы, чтобы создать свою точку зрения.
Мениппейский стиль часто требует большей продолжительности работы, чтобы создать свою точку зрения. Другой распространенный в литературе тип инструмента — пародия. Так что же означает сатира по сравнению с пародией?
Другой распространенный в литературе тип инструмента — пародия. Так что же означает сатира по сравнению с пародией?
 ,
,  и для любого
и для любого  функция f интегрируема но Риману (по Лебегу) на отрезке
функция f интегрируема но Риману (по Лебегу) на отрезке  Тогда предел
Тогда предел 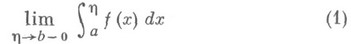
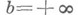 условие
условие  понимается как
понимается как  ) наз. несобственным интегралом
) наз. несобственным интегралом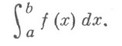
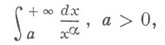 при
при  сходится, а при
сходится, а при  расходится.
Если же
расходится.
Если же  , то
, то 
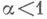 и расходится при
и расходится при  .
.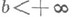 и функция f интегрируема по Риману (по Лебегу) на отрезке [ а, b], то Н. и. (1) совпадает с определенным интегралом.
и функция f интегрируема по Риману (по Лебегу) на отрезке [ а, b], то Н. и. (1) совпадает с определенным интегралом.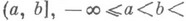
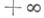
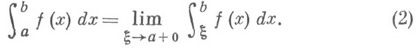
 и существуют то Н. и.
и существуют то Н. и.
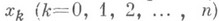 :
: 
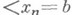 таких, что функция f интегрируема по Риману (по Лебегу) на каждом отрезке
таких, что функция f интегрируема по Риману (по Лебегу) на каждом отрезке  , не содержащем ни одной точки
, не содержащем ни одной точки 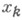 , и для каждого
, и для каждого  существуют Н. и.
существуют Н. и.

 .
. то
то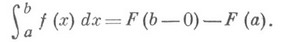
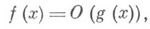
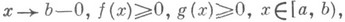 из сходимости Н. и.
из сходимости Н. и.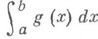
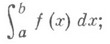
 наз. в этом случае функцией сравнения. В качестве функции сравнения для интегралов (1) в случае конечного предела интегрирования bчасто используются функции
наз. в этом случае функцией сравнения. В качестве функции сравнения для интегралов (1) в случае конечного предела интегрирования bчасто используются функции 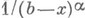 ; для интегралов вида (2) в случае конечности предела интегрирования а- функции
; для интегралов вида (2) в случае конечности предела интегрирования а- функции 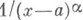 , при наличии одного или двух бесконечных пределов интегрирования — функции
, при наличии одного или двух бесконечных пределов интегрирования — функции 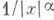 . Из признака сравнения следует, напр., если для неотрицательной функции f, определенной при
. Из признака сравнения следует, напр., если для неотрицательной функции f, определенной при  , существует предел
, существует предел
 Н. и.
Н. и.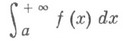
 Н. и. расходится.
Н. и. расходится. существует такое
существует такое 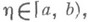 что для всех
что для всех 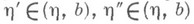 выполняется неравенство
выполняется неравенство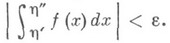 —
—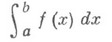


 , функция f имеет на полуоси
, функция f имеет на полуоси 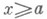 ограниченную первообразную, a g- монотонная функция, стремящаяся к нулю при
ограниченную первообразную, a g- монотонная функция, стремящаяся к нулю при  то Н. и.
то Н. и.
 , то Н. и.
, то Н. и.
 сходился ряд
сходился ряд
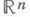 и интегрируема по Риману на любом измеримом по Жордану множестве
и интегрируема по Риману на любом измеримом по Жордану множестве  Функцию f наз. интегрируемой в несобственном смысле по множеству G, если для любой последовательности измеримых по Жордану множеств
Функцию f наз. интегрируемой в несобственном смысле по множеству G, если для любой последовательности измеримых по Жордану множеств  таких, что
таких, что 
 существует предел
существует предел 
 . Этот предел, если он существует, наз. Н. и.
. Этот предел, если он существует, наз. Н. и.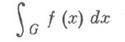

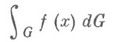
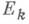 были взяты произвольные измеримые по Жордану множества. Впрочем, при
были взяты произвольные измеримые по Жордану множества. Впрочем, при 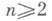 сделанное утверждение остается в силе и в том случае, когда в качестве множеств
сделанное утверждение остается в силе и в том случае, когда в качестве множеств 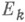 взяты только измеримые по Жордану области. Таким образом, в этом случае понятие Н. и. не приводит к новому понятию по сравнению с интегралом Лебега.
взяты только измеримые по Жордану области. Таким образом, в этом случае понятие Н. и. не приводит к новому понятию по сравнению с интегралом Лебега.
 и расходится при
и расходится при  , второй сходится при
, второй сходится при  и расходится при
и расходится при  .
. , кроме, быть может, точки
, кроме, быть может, точки  , и пусть для любого
, и пусть для любого 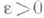 функция f интегрируема (по Риману или по Лебегу) на множестве
функция f интегрируема (по Риману или по Лебегу) на множестве 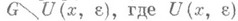 есть
есть  -окрестность точки х. Тогда если существует предел
-окрестность точки х. Тогда если существует предел 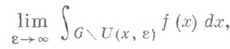

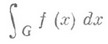
 расходится, а
расходится, а 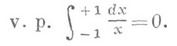
![[a,b]](/800/600/https/upload.wikimedia.org/wikipedia/commons/thumb/d/d1/Improperintegral2.png/200px-Improperintegral2.png) Несобственный интеграл первого рода
Несобственный интеграл первого рода Несобственный интеграл Римана второго рода
Несобственный интеграл Римана второго рода










 (см. Гамма-функция). К Н. и. относится и Фурье интеграл, а также интегралы, встречающиеся при др. интегральных преобразованиях. Решения краевых задач (См. Краевые задачи) математической физики записываются кратными Н. и. с неограниченной подинтегральной функцией. В теории вероятностей важное значение имеет Н. и.
(см. Гамма-функция). К Н. и. относится и Фурье интеграл, а также интегралы, встречающиеся при др. интегральных преобразованиях. Решения краевых задач (См. Краевые задачи) математической физики записываются кратными Н. и. с неограниченной подинтегральной функцией. В теории вероятностей важное значение имеет Н. и.

 В ряде случаев расходящимся Н. и. можно приписать определённое значение (см. Суммирование). В частности, если интеграл
В ряде случаев расходящимся Н. и. можно приписать определённое значение (см. Суммирование). В частности, если интеграл



 Аналогично вводится главное значение Н. и. от неограниченных функций. В работах Н. И. Мусхелишвили и его учеников построена теория интегральных уравнений, содержащих Н. и., понимаемые в смысле главного значения.
Аналогично вводится главное значение Н. и. от неограниченных функций. В работах Н. И. Мусхелишвили и его учеников построена теория интегральных уравнений, содержащих Н. и., понимаемые в смысле главного значения. для случая конечного промежутка
для случая конечного промежутка и ограниченной функции
и ограниченной функции (см. теорему 1 из §3). Теперь займемся
обобщением этого понятия для случаев
бесконечного промежутка и неограниченной
функции. Необходимость такого обобщения
показывают, например, такие ситуации.
(см. теорему 1 из §3). Теперь займемся
обобщением этого понятия для случаев
бесконечного промежутка и неограниченной
функции. Необходимость такого обобщения
показывают, например, такие ситуации. ,
, ,
то придем к интегралу от неограниченной
функции:
,
то придем к интегралу от неограниченной
функции: ,
где
,
где  .
. движется
по инерции в среде с силой сопротивления
движется
по инерции в среде с силой сопротивления  ,
где
,
где — скорость тела. Используя второй закон
Ньютона (
— скорость тела. Используя второй закон
Ньютона ( ,
где
,
где ускорение),
получим уравнение:
ускорение),
получим уравнение: ,
где
,
где .
Нетрудно показать, что решением этого
(дифференциального!) уравнения является
функция
.
Нетрудно показать, что решением этого
(дифференциального!) уравнения является
функция Если
нам потребуется вычислить путь, пройденный
телом до полной остановки, т.е. до момента,
когда
Если
нам потребуется вычислить путь, пройденный
телом до полной остановки, т.е. до момента,
когда  ,
то придем к интегралу по бесконечному
промежутку:
,
то придем к интегралу по бесконечному
промежутку:
 определена и непрерывна на промежутке
определена и непрерывна на промежутке .
Тогда для любого
.
Тогда для любого она интегрируема на промежутке
она интегрируема на промежутке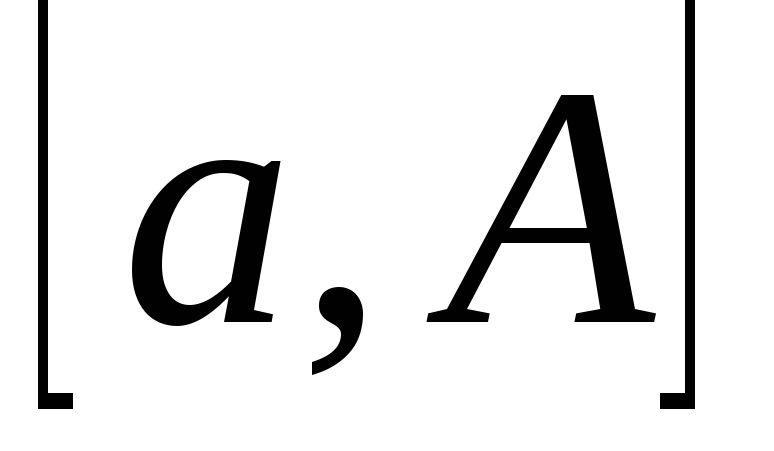 ,
то есть существует интеграл
,
то есть существует интеграл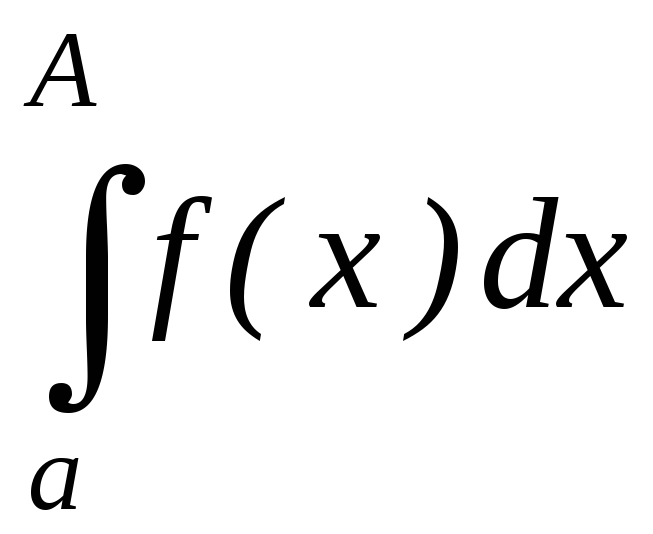 .
. называют несобственным интегралом 1-го
рода от функции
называют несобственным интегралом 1-го
рода от функции по промежутку
по промежутку и обозначают символом
и обозначают символом .
При этом, если указанный предел конечен,
то несобственный интеграл называют
сходящимся, в противном случае (
.
При этом, если указанный предел конечен,
то несобственный интеграл называют
сходящимся, в противном случае ( или не существует ) – расходящимся.
или не существует ) – расходящимся.
 .
. .
. – не существует.
– не существует. — некоторая первообразная для функции
— некоторая первообразная для функции (сущест-вует на
(сущест-вует на ,
т.к.
,
т.к. — непрерывна). Тогда
— непрерывна). Тогда
 .
Если этот предел обозначить
.
Если этот предел обозначить ,
то можно написать для интеграла (1)
формулу Ньютона-Лейбница:
,
то можно написать для интеграла (1)
формулу Ньютона-Лейбница: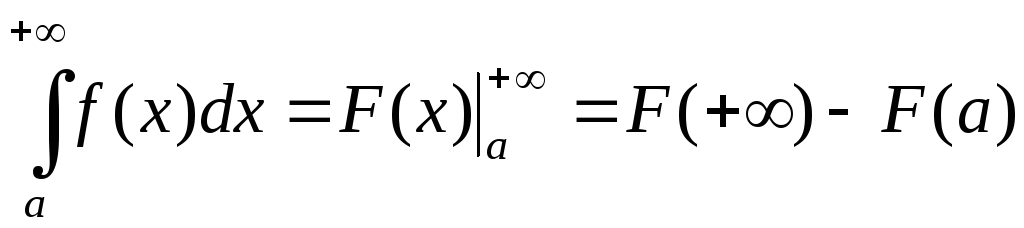 ,
где
,
где  .
.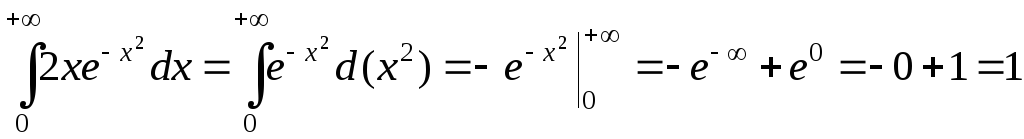 .
. .
.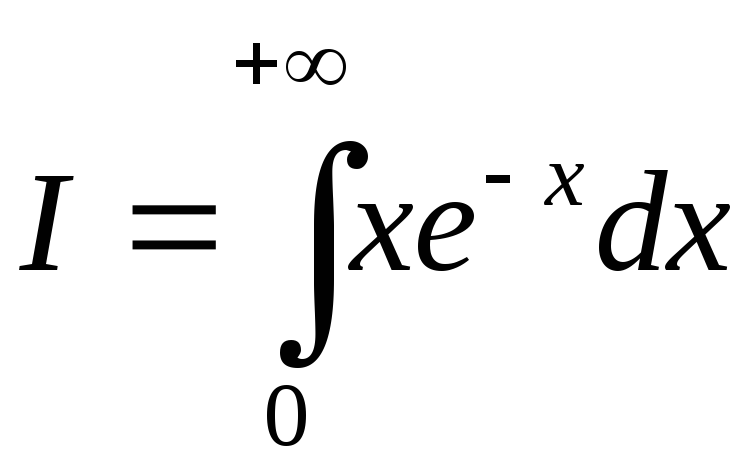 .
Сначала найдем первообразную:
.
Сначала найдем первообразную: , учитывая,
что
, учитывая,
что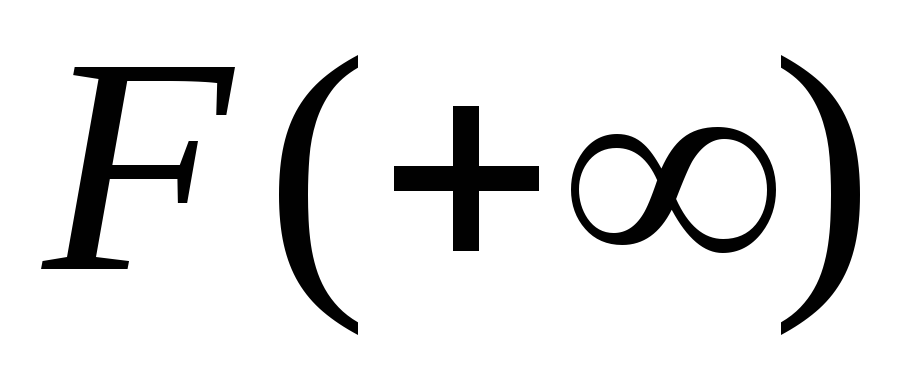
 :
: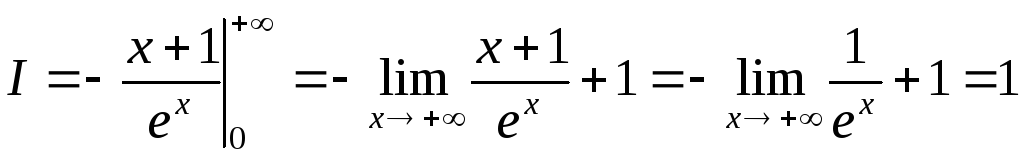 .
.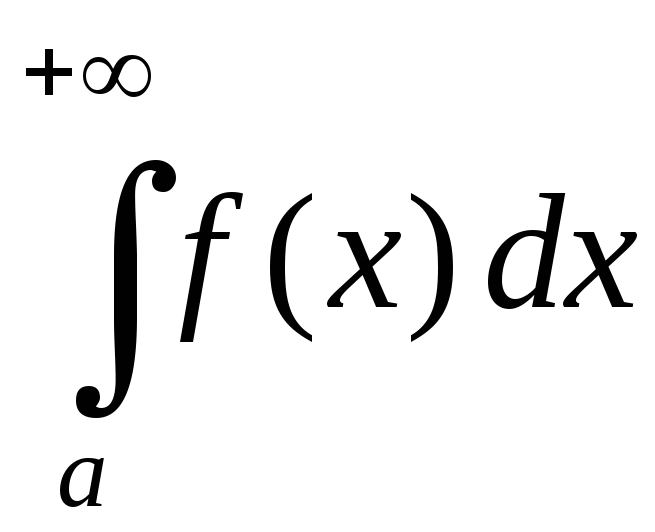 и
и
 сходятся или расходятся одновременно;
сходятся или расходятся одновременно; ,
то интегралы
,
то интегралы и
и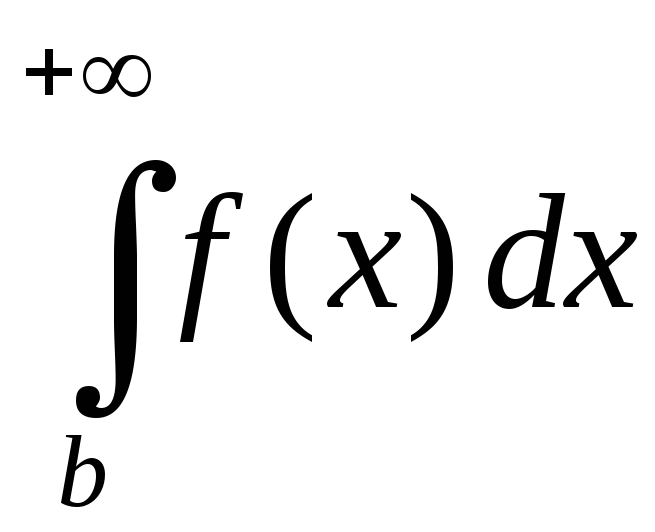 сходятся или рас-ходятся одновременно;
сходятся или рас-ходятся одновременно; сходится, то
сходится, то .
. непрерывна
на
непрерывна
на  ,
то
,
то .
. непрерывна
на
непрерывна
на ,
то принимают по определению
,
то принимают по определению (
( –
произвольное),
–
произвольное),

 (для
больших
(для
больших  ).
).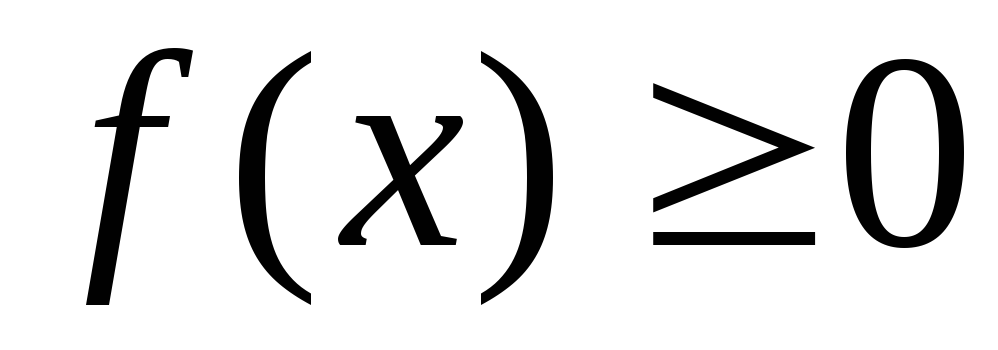 на
на  . Тогда определенный интеграл
. Тогда определенный интеграл  как функция верхнего предела есть
функция возрастаю-щая (это следует из
общих свойств определенного интеграла).
как функция верхнего предела есть
функция возрастаю-щая (это следует из
общих свойств определенного интеграла). остается
ограниченной при увеличении
остается
ограниченной при увеличении .
. и
и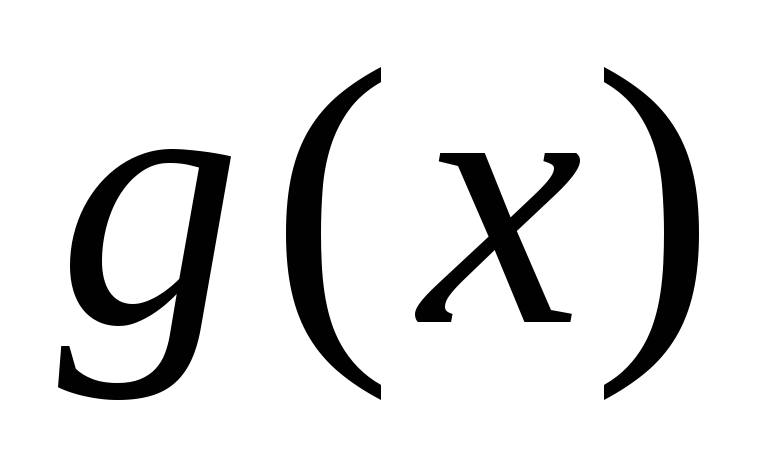 непре-рывны на
непре-рывны на и удовлетворяют неравенству
и удовлетворяют неравенству .
Тогда:
.
Тогда: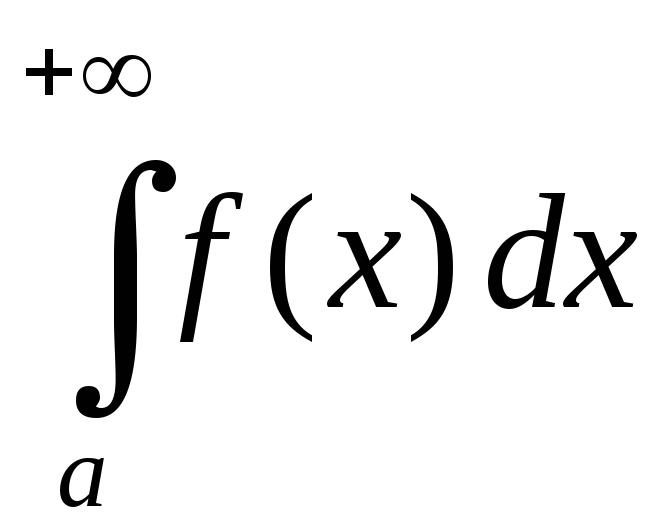 сходится;
сходится; расходится, то и
расходится, то и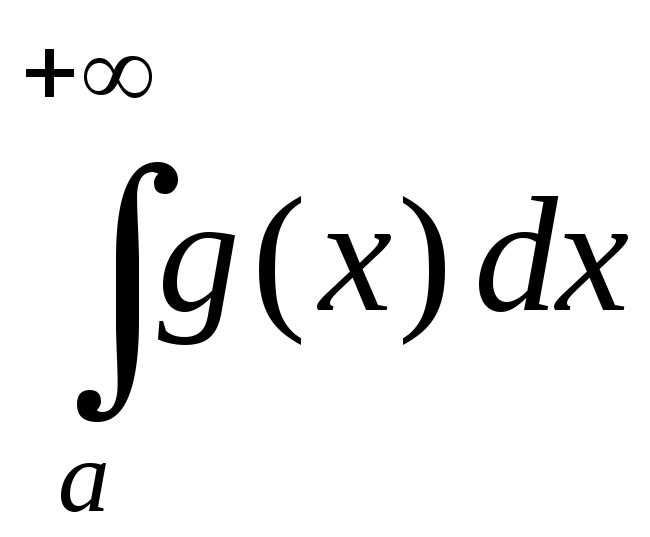 расходится.
расходится. и
и .
Так как
.
Так как ,
то
,
то
 .
Пусть интеграл
.
Пусть интеграл сходится, тогда (в силу теоремы 1) функция
сходится, тогда (в силу теоремы 1) функция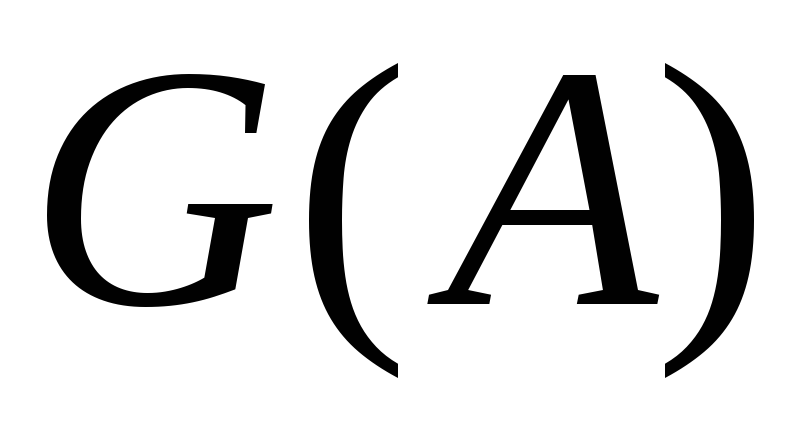 ‒ ограничена. Но тогда и
‒ ограничена. Но тогда и ограничена,
а значит, интеграл
ограничена,
а значит, интеграл тоже сходится. Аналогично доказывается
и вторая часть теоремы.
тоже сходится. Аналогично доказывается
и вторая часть теоремы. или сходимости интеграла от
или сходимости интеграла от .
Этот недостаток отсутствует у 2-го
признака сравнения.
.
Этот недостаток отсутствует у 2-го
признака сравнения. и
и непрерывны и неотрицательны на
непрерывны и неотрицательны на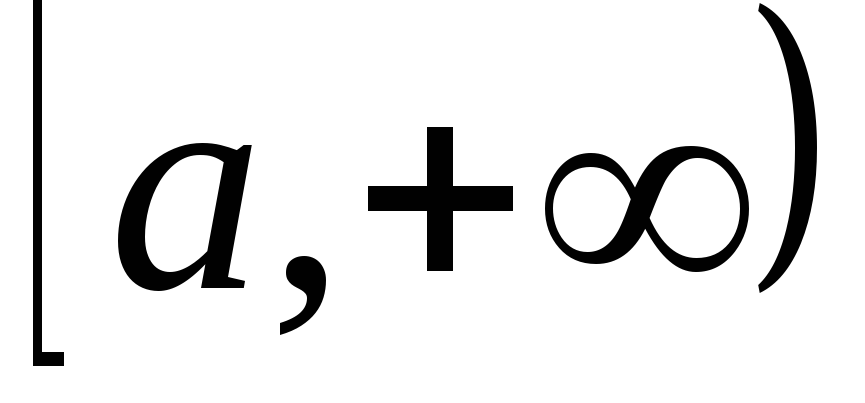 .
Тогда, если
.
Тогда, если при
при ,
то несобственные интегралы
,
то несобственные интегралы и
и сходятся или расходятся одновременно.
сходятся или расходятся одновременно. ,
, 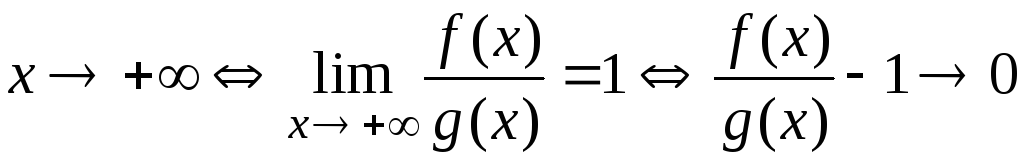 ,
,
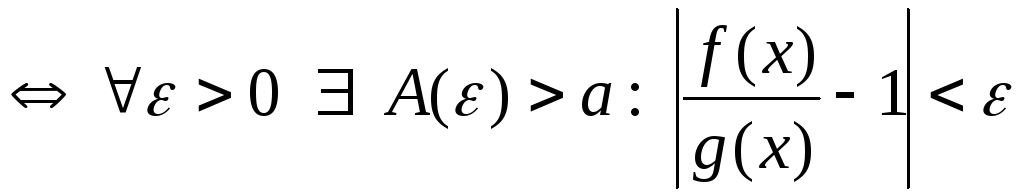
 .
. .
Тогда:
.
Тогда: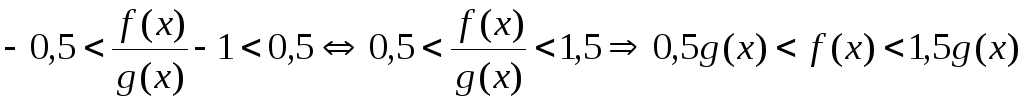 .
.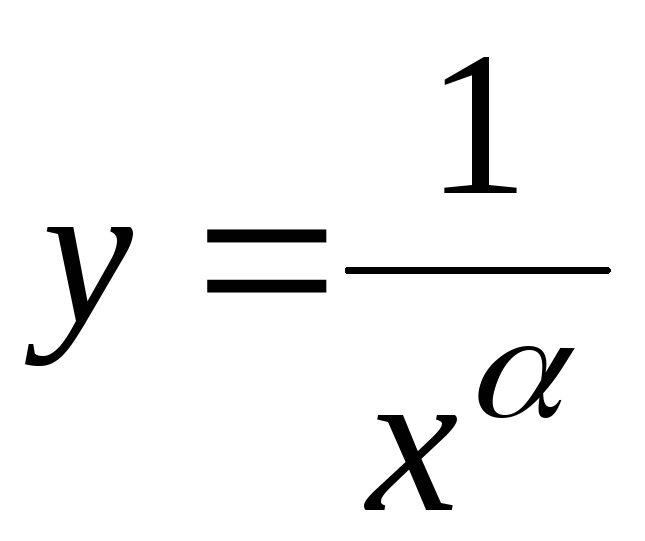 ,
, .
Предлагаем студентам самим доказать,
что интеграл
.
Предлагаем студентам самим доказать,
что интеграл
 и расходится при
и расходится при .
. .
. :
: ,
,  .
.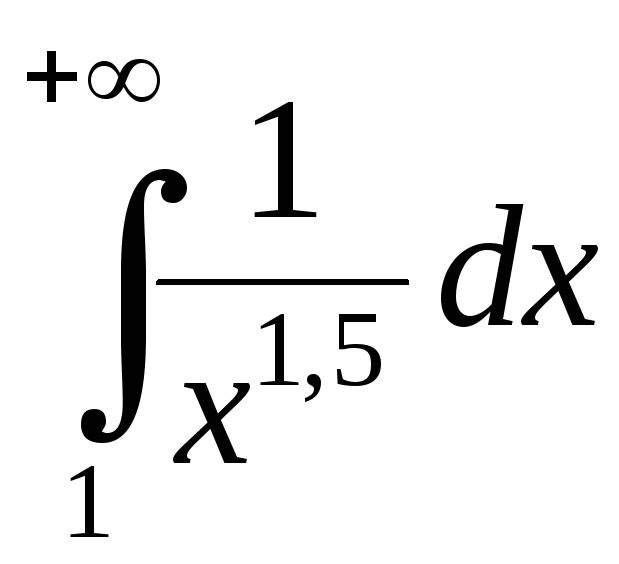 сходится, ибо
сходится, ибо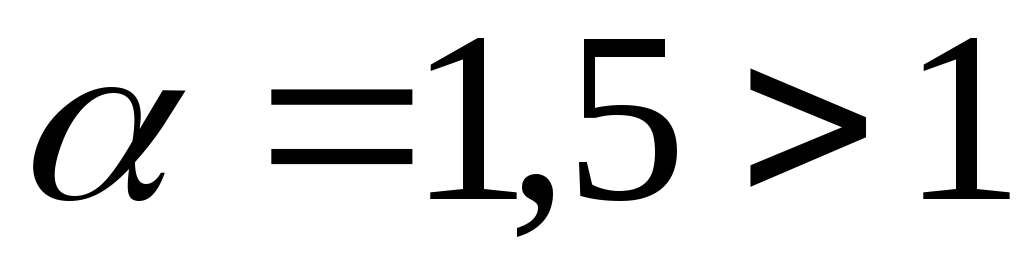 .
По 2-му признаку сравнения сходится и
интеграл
.
По 2-му признаку сравнения сходится и
интеграл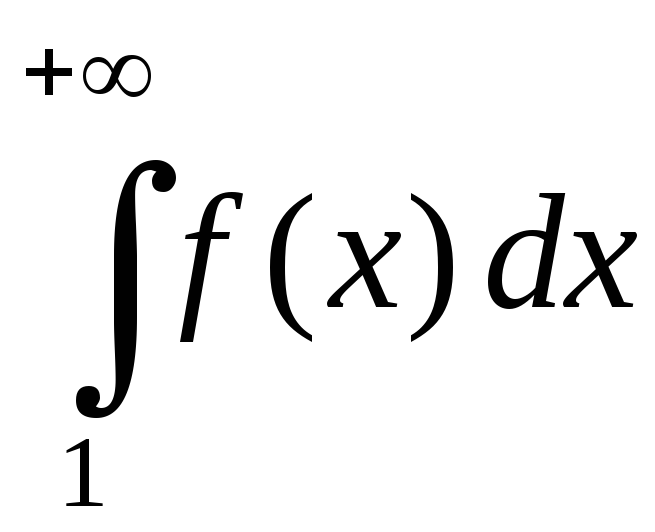 ,
а в силу свойства 2) из §1 сходится и
исход-ный интеграл.
,
а в силу свойства 2) из §1 сходится и
исход-ный интеграл. .
.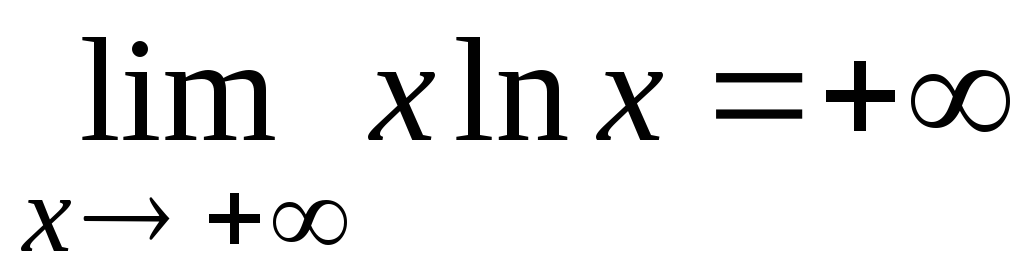 ,
тоcуществует
,
тоcуществует  такое, что при
такое, что при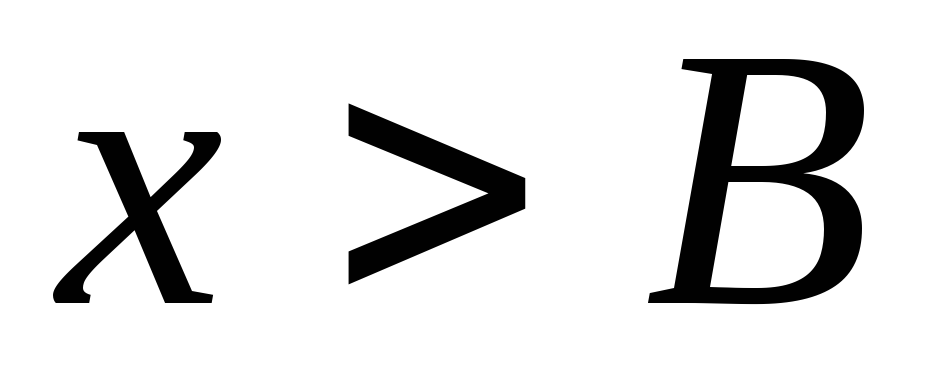
 .
Для таких значений переменной:
.
Для таких значений переменной: .
. ,
, .
. сходится как эталонный. В силу 1-го
признака сравнения сходится и
сходится как эталонный. В силу 1-го
признака сравнения сходится и .
Применяя 2-й признак, получим, что и
интеграл
.
Применяя 2-й признак, получим, что и
интеграл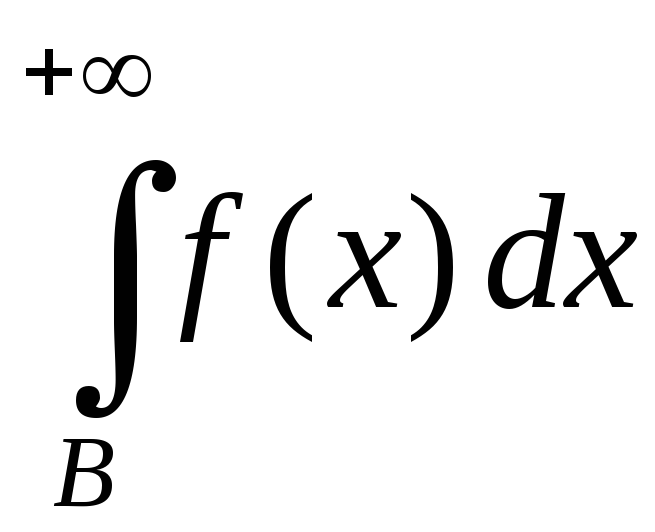 сходится. И снова свойство 2) из §1
доказывает сходимость исходного
интеграла.
сходится. И снова свойство 2) из §1
доказывает сходимость исходного
интеграла. ru).
Мы готовы устранить любые неудобства, связанные с использованием данной таблицы, или ей подобных, которые можно найти в разделе
«Физика».
ru).
Мы готовы устранить любые неудобства, связанные с использованием данной таблицы, или ей подобных, которые можно найти в разделе
«Физика». Молекулярная физика
Молекулярная физика Направление сформировалось несколько позднее, чем статика и динамика: во второй половине XIX столетия. Первые исследования в области кинематики были посвящены огнестрельному оружию. Ученые стремились понять процесс полета снаряда, производили расчет траектории его движения. В дальнейшем кинематика как научное направление получило широкое распространение и существенно повлияло на развитие технического прогресса.
Направление сформировалось несколько позднее, чем статика и динамика: во второй половине XIX столетия. Первые исследования в области кинематики были посвящены огнестрельному оружию. Ученые стремились понять процесс полета снаряда, производили расчет траектории его движения. В дальнейшем кинематика как научное направление получило широкое распространение и существенно повлияло на развитие технического прогресса. При рассмотрении замкнутой траектории перемещение будет равно нулю.
При рассмотрении замкнутой траектории перемещение будет равно нулю.
 {2}}\)
{2}}\) {2}}=\frac{9,81}{0,17}=57,7\)
{2}}=\frac{9,81}{0,17}=57,7\)


 Значение этого слова подразумевает под собой изменение чего-либо. Например, политическое движение выступает за равноправие разных слоев населения вне зависимости от их расовой принадлежности. Раньше его не было, затем что-то изменилось и теперь каждый человек имеет равные права. Это движение цивилизации вперед. Еще пример — экологическое. В прошлом, выбравшись на природу, никто не задумывался о том, что оставляет после себя мусор. Сегодня же любой цивилизованный человек соберет его за собой и отвезет в специально отведенное место для дальнейшей утилизации.
Значение этого слова подразумевает под собой изменение чего-либо. Например, политическое движение выступает за равноправие разных слоев населения вне зависимости от их расовой принадлежности. Раньше его не было, затем что-то изменилось и теперь каждый человек имеет равные права. Это движение цивилизации вперед. Еще пример — экологическое. В прошлом, выбравшись на природу, никто не задумывался о том, что оставляет после себя мусор. Сегодня же любой цивилизованный человек соберет его за собой и отвезет в специально отведенное место для дальнейшей утилизации.
 Большего количества чисел в трехмерном мире (для описывания положения материальной точки) не требуется.
Большего количества чисел в трехмерном мире (для описывания положения материальной точки) не требуется. Расстояние, пройденное грузовиком, составляет 110 м. Найти ускорение.
Расстояние, пройденное грузовиком, составляет 110 м. Найти ускорение. Такое сложное движение светил, связанное с тем, что земные наблюдатели находятся в системе отсчета, которая очень замысловато движется. Земля вращается вокруг свое оси и одновременно вокруг Солнца. На самом деле, если сменить систему отсчета, то все движения небесных тел легко описываются. Это в свое время было сделано Коперником. Он предложил собственное описание мироустройства, в котором Солнце неподвижно. Относительно него описать движение планет гораздо проще, чем если телом отсчета будет являться Земля.
Такое сложное движение светил, связанное с тем, что земные наблюдатели находятся в системе отсчета, которая очень замысловато движется. Земля вращается вокруг свое оси и одновременно вокруг Солнца. На самом деле, если сменить систему отсчета, то все движения небесных тел легко описываются. Это в свое время было сделано Коперником. Он предложил собственное описание мироустройства, в котором Солнце неподвижно. Относительно него описать движение планет гораздо проще, чем если телом отсчета будет являться Земля. Их и возьмем!
Их и возьмем! Это я делаю только для того, чтобы Вам легче было читать. Но всегда помните, что речь идет о геометрической точке.
Это я делаю только для того, чтобы Вам легче было читать. Но всегда помните, что речь идет о геометрической точке.


 Система отсчета (СО) – совокупность тела отсчета (оно считается абсолютно твердым), привязанной к нему системой координат, линейки (прибора, измеряющего расстояния), часов и синхронизатора времени.
Система отсчета (СО) – совокупность тела отсчета (оно считается абсолютно твердым), привязанной к нему системой координат, линейки (прибора, измеряющего расстояния), часов и синхронизатора времени. При этом не забывайте, что полное перемещение не всегда равно алгебраической сумме перемещений на определённых этапах движения. Вектор полного перемещения равен векторной сумме перемещений на отдельных этапах движения.
При этом не забывайте, что полное перемещение не всегда равно алгебраической сумме перемещений на определённых этапах движения. Вектор полного перемещения равен векторной сумме перемещений на отдельных этапах движения. При равноускоренном движении среднюю скорость можно рассчитывать, как среднее арифметическое начальной и конечной скоростей (этим свойством очень удобно пользоваться при решении некоторых задач):
При равноускоренном движении среднюю скорость можно рассчитывать, как среднее арифметическое начальной и конечной скоростей (этим свойством очень удобно пользоваться при решении некоторых задач): Смысл этого индекса и правило определения знаков сохраняется. Куда направлять ось OY – Ваш выбор, зависящий от удобства решения задачи. Вариантов 2: вверх или вниз.
Смысл этого индекса и правило определения знаков сохраняется. Куда направлять ось OY – Ваш выбор, зависящий от удобства решения задачи. Вариантов 2: вверх или вниз. Тогда этот угол будет находиться из соотношения:
Тогда этот угол будет находиться из соотношения: Таким образом, покой и движение тела относительны.
Таким образом, покой и движение тела относительны. Не забывайте переводить углы из градусов в радианы. Длина дуги l связана с углом поворота соотношением:
Не забывайте переводить углы из градусов в радианы. Длина дуги l связана с углом поворота соотношением: Также в ходе РТ важно привыкнуть к стилю постановки вопросов в задачах, который на ЦТ может показаться неподготовленному человеку очень непривычным.
Также в ходе РТ важно привыкнуть к стилю постановки вопросов в задачах, который на ЦТ может показаться неподготовленному человеку очень непривычным.
 Они являются идеальными моделями материальных тел с той или иной степенью абстракции конкретных свойств реальных физических тел.
Они являются идеальными моделями материальных тел с той или иной степенью абстракции конкретных свойств реальных физических тел.
 2}$
2}$


 Однако вы не знаете, какое смещение испытает ваша машина, если бы вы резко нажали на тормоз и занесло до полной остановки; и вы не знаете, сколько времени потребуется, чтобы остановиться. В таком случае неизвестные параметры могут быть определены с использованием физических принципов и математических уравнений (кинематических уравнений).
Однако вы не знаете, какое смещение испытает ваша машина, если бы вы резко нажали на тормоз и занесло до полной остановки; и вы не знаете, сколько времени потребуется, чтобы остановиться. В таком случае неизвестные параметры могут быть определены с использованием физических принципов и математических уравнений (кинематических уравнений).

 Таким образом, вы будете искать уравнение, в котором перечислены эти четыре переменные. Анализ четырех приведенных выше уравнений показывает, что уравнение в правом верхнем углу содержит все четыре переменные.
Таким образом, вы будете искать уравнение, в котором перечислены эти четыре переменные. Анализ четырех приведенных выше уравнений показывает, что уравнение в правом верхнем углу содержит все четыре переменные. Когда он наконец стал зеленым, Бен ускорился из состояния покоя со скоростью 6,00 м / с 2 за время 4,10 секунды. Определите перемещение машины Бена за этот период времени.
Когда он наконец стал зеленым, Бен ускорился из состояния покоя со скоростью 6,00 м / с 2 за время 4,10 секунды. Определите перемещение машины Бена за этот период времени. Этот шаг показан ниже.
Этот шаг показан ниже.
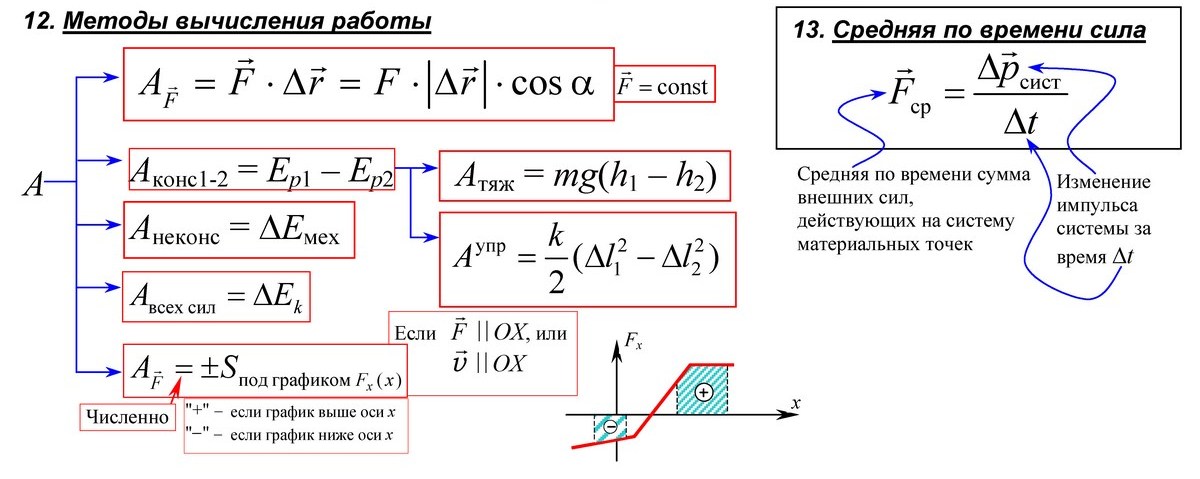 Это намного эффективнее, чем запоминание списка фактов. Аналитические навыки и способности решать проблемы могут быть применены к новым ситуациям, тогда как список фактов не может быть достаточно длинным, чтобы содержать все возможные обстоятельства. Такие аналитические навыки полезны как для решения задач на уроках физики, так и для применения физики в повседневной и профессиональной жизни.
Это намного эффективнее, чем запоминание списка фактов. Аналитические навыки и способности решать проблемы могут быть применены к новым ситуациям, тогда как список фактов не может быть достаточно длинным, чтобы содержать все возможные обстоятельства. Такие аналитические навыки полезны как для решения задач на уроках физики, так и для применения физики в повседневной и профессиональной жизни. По этой причине диаграмма движения дает больше информации, чем диаграмма пути. Он также может отображать силы, действующие на объект в каждый момент времени.
По этой причине диаграмма движения дает больше информации, чем диаграмма пути. Он также может отображать силы, действующие на объект в каждый момент времени.
 0 м / с 2 от полной остановки, сколько времени потребуется, чтобы проехать 3000 м?
0 м / с 2 от полной остановки, сколько времени потребуется, чтобы проехать 3000 м? Однако не всегда такая полнота известна.Часто бывает так, что известны лишь некоторые параметры движения объекта, а остальные неизвестны. Например, приближаясь к светофору, вы можете узнать, что ваша машина имеет скорость 22 м / с, восток, и способна к заносу 8,0 м / с 2 , запад.
Однако не всегда такая полнота известна.Часто бывает так, что известны лишь некоторые параметры движения объекта, а остальные неизвестны. Например, приближаясь к светофору, вы можете узнать, что ваша машина имеет скорость 22 м / с, восток, и способна к заносу 8,0 м / с 2 , запад. Каждый символ имеет свое особое значение. Символ d обозначает смещение объекта.
Каждый символ имеет свое особое значение. Символ d обозначает смещение объекта.
 Каждый символ имеет свое особое значение. Символ d обозначает смещение объекта. Символ t обозначает время, в течение которого объект двигался. Символ обозначает ускорение объекта.А символ v обозначает скорость объекта; индекс i после v (как в v i ) указывает, что значение скорости является начальным значением скорости, а индекс f (как в v f ) указывает, что значение скорости является конечным значением скорости.
Каждый символ имеет свое особое значение. Символ d обозначает смещение объекта. Символ t обозначает время, в течение которого объект двигался. Символ обозначает ускорение объекта.А символ v обозначает скорость объекта; индекс i после v (как в v i ) указывает, что значение скорости является начальным значением скорости, а индекс f (как в v f ) указывает, что значение скорости является конечным значением скорости. Учитывая, что такое название было бы стилистическим кошмаром, позвольте мне начать этот раздел со следующей оговорки. Эти уравнения движения действительны только тогда, когда ускорение постоянное и движение ограничено прямой линией.
Учитывая, что такое название было бы стилистическим кошмаром, позвольте мне начать этот раздел со следующей оговорки. Эти уравнения движения действительны только тогда, когда ускорение постоянное и движение ограничено прямой линией. В этом порядке их также часто называют первым, вторым и третьим уравнениями движения, но нет веских причин для изучения этих имен.
В этом порядке их также часто называют первым, вторым и третьим уравнениями движения, но нет веских причин для изучения этих имен. Его часто называют «первой скоростью», но это довольно наивный способ Опишите это. Лучшее определение было бы сказать, что начальная скорость — это скорость, которую имеет движущийся объект, когда он впервые становится важным в проблеме. Скажем, метеор был замечен глубоко в космосе, и проблема заключалась в том, чтобы определить его траекторию, тогда начальная скорость, вероятно, будет той скоростью, которую он имел при первом наблюдении.Но если проблема заключалась в том, что тот же самый метеор сгорает при входе в атмосферу, то начальная скорость, вероятно, равна скорости, которую он имел при входе в атмосферу Земли. Ответ на вопрос «Какая начальная скорость?» «Это зависит от обстоятельств». Это оказывается ответом на множество вопросов.
Его часто называют «первой скоростью», но это довольно наивный способ Опишите это. Лучшее определение было бы сказать, что начальная скорость — это скорость, которую имеет движущийся объект, когда он впервые становится важным в проблеме. Скажем, метеор был замечен глубоко в космосе, и проблема заключалась в том, чтобы определить его траекторию, тогда начальная скорость, вероятно, будет той скоростью, которую он имел при первом наблюдении.Но если проблема заключалась в том, что тот же самый метеор сгорает при входе в атмосферу, то начальная скорость, вероятно, равна скорости, которую он имел при входе в атмосферу Земли. Ответ на вопрос «Какая начальная скорость?» «Это зависит от обстоятельств». Это оказывается ответом на множество вопросов. Если бы он стартовал со скоростью 15 м / с, то его скорость через 5 с была бы…
Если бы он стартовал со скоростью 15 м / с, то его скорость через 5 с была бы…
 Символ s — это позиция на t позже. Если хотите, вы можете называть ее конечной позицией . Изменение положения (∆ s ) называется смещением или расстоянием (в зависимости от обстоятельств), и некоторые люди предпочитают писать второе уравнение движения таким образом.
Символ s — это позиция на t позже. Если хотите, вы можете называть ее конечной позицией . Изменение положения (∆ s ) называется смещением или расстоянием (в зависимости от обстоятельств), и некоторые люди предпочитают писать второе уравнение движения таким образом.

 Выполните эту операцию в обратном порядке. Вместо того, чтобы различать положение для определения скорости, интегрируйте скорость, чтобы найти положение. Это дает нам уравнение положения-времени для постоянного ускорения, также известное как второе уравнение движения [2].
Выполните эту операцию в обратном порядке. Вместо того, чтобы различать положение для определения скорости, интегрируйте скорость, чтобы найти положение. Это дает нам уравнение положения-времени для постоянного ускорения, также известное как второе уравнение движения [2]. Это произошло от этой производной…
Это произошло от этой производной… Однако на самом деле это сработало только потому, что ускорение было постоянным — постоянным во времени и постоянным в пространстве. Если бы ускорение каким-либо образом менялось, этот метод был бы неудобно трудным. Мы вернемся к алгебре, чтобы спасти наше здравомыслие. Не то чтобы в этом что-то не так. Алгебра работает, а здравомыслие стоит сэкономить.
Однако на самом деле это сработало только потому, что ускорение было постоянным — постоянным во времени и постоянным в пространстве. Если бы ускорение каким-либо образом менялось, этот метод был бы неудобно трудным. Мы вернемся к алгебре, чтобы спасти наше здравомыслие. Не то чтобы в этом что-то не так. Алгебра работает, а здравомыслие стоит сэкономить. Он представил, что положение увеличивается с квадратом времени, что часто называют Законом падающих тел. Последний пункт в этом отрывке, который он представил, заключается в том, что скорость увеличивается с квадратом расстояния вниз по рампе.
Он представил, что положение увеличивается с квадратом времени, что часто называют Законом падающих тел. Последний пункт в этом отрывке, который он представил, заключается в том, что скорость увеличивается с квадратом расстояния вниз по рампе. Это уравнение еще больше упрощается и становится
Это уравнение еще больше упрощается и становится










 Хорошие манеры – это отличительная черта умных людей. Но какие манеры хорошие, а какие плохие? Речевой этикет рассказывает о хороших манерах в речи, которые помогут уверенно общаться с людьми.
Хорошие манеры – это отличительная черта умных людей. Но какие манеры хорошие, а какие плохие? Речевой этикет рассказывает о хороших манерах в речи, которые помогут уверенно общаться с людьми. Скачать PDF
Скачать PDF Распечатать
Распечатать
 Снова противоречие.
Снова противоречие.
 Горожанин Марк выкрикнул: « Или я лжец, или Артур рыцарь!». Кто такой Марк (рыцарь или лжец), и кто такой Артур?
Горожанин Марк выкрикнул: « Или я лжец, или Артур рыцарь!». Кто такой Марк (рыцарь или лжец), и кто такой Артур?


 То есть Сем – лжец. Значит, они оба лжецы. Но тогда обе части высказывания Тома истинны, что невозможно, следовательно, Том – лжец). Значит, этот город не может называться Троя. Получается, что Том лжец, а Сем – рыцарь.
То есть Сем – лжец. Значит, они оба лжецы. Но тогда обе части высказывания Тома истинны, что невозможно, следовательно, Том – лжец). Значит, этот город не может называться Троя. Получается, что Том лжец, а Сем – рыцарь.
 Потом каждого спросили: «Делится ли ваше число на 2?». Утвердительный ответ дали 3 человека. На вопрос «Делится ли ваше число на 4?» утвердительный ответ дали 6 человек. На вопрос «Делится ли ваше число на 5?» утвердительно ответили 2 человека. Сколько было лжецов и какие у них были числа?
Потом каждого спросили: «Делится ли ваше число на 2?». Утвердительный ответ дали 3 человека. На вопрос «Делится ли ваше число на 4?» утвердительный ответ дали 6 человек. На вопрос «Делится ли ваше число на 5?» утвердительно ответили 2 человека. Сколько было лжецов и какие у них были числа? Рыцари говорят только правду, а лжецы всегда лгут. У каждого рыцаря есть хотя бы один друг, а у каждого лжеца ровно один друг. Как-то раз каждый произнес фразу «Ни один из моих друзей не является лжецом», либо «Ни один из моих друзей не является рыцарем», причем каждую из фраз произнесло ровно 50 человек. Какое наименьшее возможное число пар друзей, один из которых рыцарь, а другой — лжец?
Рыцари говорят только правду, а лжецы всегда лгут. У каждого рыцаря есть хотя бы один друг, а у каждого лжеца ровно один друг. Как-то раз каждый произнес фразу «Ни один из моих друзей не является лжецом», либо «Ни один из моих друзей не является рыцарем», причем каждую из фраз произнесло ровно 50 человек. Какое наименьшее возможное число пар друзей, один из которых рыцарь, а другой — лжец? Каждый из них заявил остальным: «Вы все – лжецы!» Сколько рыцарей было среди этих тринадцати жителей?
Каждый из них заявил остальным: «Вы все – лжецы!» Сколько рыцарей было среди этих тринадцати жителей? В одной супружеской паре мистер Хек высказал следующие утверждения: «Моя жена — не хитрец», а миссис Хек заявила: «Мой муж — не хитрец». Кто такой мистер Хек и кто такая миссис Хек — рыцарь, лжец или хитрец?
В одной супружеской паре мистер Хек высказал следующие утверждения: «Моя жена — не хитрец», а миссис Хек заявила: «Мой муж — не хитрец». Кто такой мистер Хек и кто такая миссис Хек — рыцарь, лжец или хитрец? Оборотнем может быть, как рыцарь, так и лжец. Знакомый рыцарь вам шепнул, что один из претендентов точно оборотень. Первый претендент заявил вам: «Оборотень – рыцарь». Второй сказал: «Оборотень – лжец». Кого вы возьмете в проводники?
Оборотнем может быть, как рыцарь, так и лжец. Знакомый рыцарь вам шепнул, что один из претендентов точно оборотень. Первый претендент заявил вам: «Оборотень – рыцарь». Второй сказал: «Оборотень – лжец». Кого вы возьмете в проводники? А заявил, что Б лжец. Но преступление совершил В. Б заявил, что А и В либо оба рыцари, либо оба лжецы. В высказался, что Б говорит правду. Но тем не менее он и совершил преступление. Помогите судье определить кто есть кто (лжецы и рыцари), и кто совершил преступление.
А заявил, что Б лжец. Но преступление совершил В. Б заявил, что А и В либо оба рыцари, либо оба лжецы. В высказался, что Б говорит правду. Но тем не менее он и совершил преступление. Помогите судье определить кто есть кто (лжецы и рыцари), и кто совершил преступление. lst-kix_list_7-7{list-style-type:none}#doc18949057 ol.lst-kix_list_7-8{list-style-type:none}#doc18949057 ol.lst-kix_list_7-1{list-style-type:none}#doc18949057 ol.lst-kix_list_1-5.start{counter-reset:lst-ctn-kix_list_1-5 0}#doc18949057 ol.lst-kix_list_7-2{list-style-type:none}#doc18949057 ol.lst-kix_list_7-3{list-style-type:none}#doc18949057 ol.lst-kix_list_7-4{list-style-type:none}#doc18949057 ol.lst-kix_list_5-3.start{counter-reset:lst-ctn-kix_list_5-3 0}#doc18949057 .lst-kix_list_2-3>li{counter-increment:lst-ctn-kix_list_2-3}#doc18949057 .lst-kix_list_4-3>li{counter-increment:lst-ctn-kix_list_4-3}#doc18949057 ol.lst-kix_list_4-5.start{counter-reset:lst-ctn-kix_list_4-5 0}#doc18949057 .lst-kix_list_1-2>li{counter-increment:lst-ctn-kix_list_1-2}#doc18949057 ol.lst-kix_list_3-7.start{counter-reset:lst-ctn-kix_list_3-7 0}#doc18949057 .lst-kix_list_5-2>li{counter-increment:lst-ctn-kix_list_5-2}#doc18949057 .lst-kix_list_3-2>li{counter-increment:lst-ctn-kix_list_3-2}#doc18949057 .lst-kix_list_7-2>li{counter-increment:lst-ctn-kix_list_7-2}#doc18949057 .lst-kix_list_5-0>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «. «}#doc18949057 ol.lst-kix_list_6-0{list-style-type:none}#doc18949057 ol.lst-kix_list_6-1{list-style-type:none}#doc18949057 .lst-kix_list_5-4>li{counter-increment:lst-ctn-kix_list_5-4}#doc18949057 .lst-kix_list_1-4>li{counter-increment:lst-ctn-kix_list_1-4}#doc18949057 ol.lst-kix_list_1-6.start{counter-reset:lst-ctn-kix_list_1-6 0}#doc18949057 .lst-kix_list_5-3>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «. «}#doc18949057 .lst-kix_list_5-2>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «. «}#doc18949057 .lst-kix_list_5-1>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.
lst-kix_list_7-7{list-style-type:none}#doc18949057 ol.lst-kix_list_7-8{list-style-type:none}#doc18949057 ol.lst-kix_list_7-1{list-style-type:none}#doc18949057 ol.lst-kix_list_1-5.start{counter-reset:lst-ctn-kix_list_1-5 0}#doc18949057 ol.lst-kix_list_7-2{list-style-type:none}#doc18949057 ol.lst-kix_list_7-3{list-style-type:none}#doc18949057 ol.lst-kix_list_7-4{list-style-type:none}#doc18949057 ol.lst-kix_list_5-3.start{counter-reset:lst-ctn-kix_list_5-3 0}#doc18949057 .lst-kix_list_2-3>li{counter-increment:lst-ctn-kix_list_2-3}#doc18949057 .lst-kix_list_4-3>li{counter-increment:lst-ctn-kix_list_4-3}#doc18949057 ol.lst-kix_list_4-5.start{counter-reset:lst-ctn-kix_list_4-5 0}#doc18949057 .lst-kix_list_1-2>li{counter-increment:lst-ctn-kix_list_1-2}#doc18949057 ol.lst-kix_list_3-7.start{counter-reset:lst-ctn-kix_list_3-7 0}#doc18949057 .lst-kix_list_5-2>li{counter-increment:lst-ctn-kix_list_5-2}#doc18949057 .lst-kix_list_3-2>li{counter-increment:lst-ctn-kix_list_3-2}#doc18949057 .lst-kix_list_7-2>li{counter-increment:lst-ctn-kix_list_7-2}#doc18949057 .lst-kix_list_5-0>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «. «}#doc18949057 ol.lst-kix_list_6-0{list-style-type:none}#doc18949057 ol.lst-kix_list_6-1{list-style-type:none}#doc18949057 .lst-kix_list_5-4>li{counter-increment:lst-ctn-kix_list_5-4}#doc18949057 .lst-kix_list_1-4>li{counter-increment:lst-ctn-kix_list_1-4}#doc18949057 ol.lst-kix_list_1-6.start{counter-reset:lst-ctn-kix_list_1-6 0}#doc18949057 .lst-kix_list_5-3>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «. «}#doc18949057 .lst-kix_list_5-2>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «. «}#doc18949057 .lst-kix_list_5-1>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «. «}#doc18949057 .lst-kix_list_5-7>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «.» counter(lst-ctn-kix_list_5-5,decimal) «.» counter(lst-ctn-kix_list_5-6,decimal) «.» counter(lst-ctn-kix_list_5-7,decimal) «. «}#doc18949057 .lst-kix_list_5-6>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «.» counter(lst-ctn-kix_list_5-5,decimal) «.» counter(lst-ctn-kix_list_5-6,decimal) «. «}#doc18949057 .lst-kix_list_5-8>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «.» counter(lst-ctn-kix_list_5-5,decimal) «.» counter(lst-ctn-kix_list_5-6,decimal) «.» counter(lst-ctn-kix_list_5-7,decimal) «.» counter(lst-ctn-kix_list_5-8,decimal) «. «}#doc18949057 ol.lst-kix_list_6-6{list-style-type:none}#doc18949057 ol.lst-kix_list_6-7{list-style-type:none}#doc18949057 .lst-kix_list_5-4>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «. «}#doc18949057 ol.lst-kix_list_6-8{list-style-type:none}#doc18949057 .lst-kix_list_5-5>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «.» counter(lst-ctn-kix_list_5-5,decimal) «. «}#doc18949057 ol.lst-kix_list_6-2{list-style-type:none}#doc18949057 ol.lst-kix_list_6-3{list-style-type:none}#doc18949057 ol.
«}#doc18949057 .lst-kix_list_5-7>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «.» counter(lst-ctn-kix_list_5-5,decimal) «.» counter(lst-ctn-kix_list_5-6,decimal) «.» counter(lst-ctn-kix_list_5-7,decimal) «. «}#doc18949057 .lst-kix_list_5-6>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «.» counter(lst-ctn-kix_list_5-5,decimal) «.» counter(lst-ctn-kix_list_5-6,decimal) «. «}#doc18949057 .lst-kix_list_5-8>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «.» counter(lst-ctn-kix_list_5-5,decimal) «.» counter(lst-ctn-kix_list_5-6,decimal) «.» counter(lst-ctn-kix_list_5-7,decimal) «.» counter(lst-ctn-kix_list_5-8,decimal) «. «}#doc18949057 ol.lst-kix_list_6-6{list-style-type:none}#doc18949057 ol.lst-kix_list_6-7{list-style-type:none}#doc18949057 .lst-kix_list_5-4>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «. «}#doc18949057 ol.lst-kix_list_6-8{list-style-type:none}#doc18949057 .lst-kix_list_5-5>li:before{content:»» counter(lst-ctn-kix_list_5-0,decimal) «.» counter(lst-ctn-kix_list_5-1,decimal) «.» counter(lst-ctn-kix_list_5-2,decimal) «.» counter(lst-ctn-kix_list_5-3,decimal) «.» counter(lst-ctn-kix_list_5-4,decimal) «.» counter(lst-ctn-kix_list_5-5,decimal) «. «}#doc18949057 ol.lst-kix_list_6-2{list-style-type:none}#doc18949057 ol.lst-kix_list_6-3{list-style-type:none}#doc18949057 ol. lst-kix_list_6-4{list-style-type:none}#doc18949057 ol.lst-kix_list_6-5{list-style-type:none}#doc18949057 ol.lst-kix_list_1-0.start{counter-reset:lst-ctn-kix_list_1-0 0}#doc18949057 .lst-kix_list_6-1>li:before{content:»» counter(lst-ctn-kix_list_6-1,lower-latin) «. «}#doc18949057 .lst-kix_list_6-3>li:before{content:»» counter(lst-ctn-kix_list_6-3,decimal) «. «}#doc18949057 .lst-kix_list_6-5>li{counter-increment:lst-ctn-kix_list_6-5}#doc18949057 .lst-kix_list_6-8>li{counter-increment:lst-ctn-kix_list_6-8}#doc18949057 .lst-kix_list_6-0>li:before{content:»» counter(lst-ctn-kix_list_6-0,decimal) «. «}#doc18949057 .lst-kix_list_6-4>li:before{content:»» counter(lst-ctn-kix_list_6-4,lower-latin) «. «}#doc18949057 .lst-kix_list_3-0>li{counter-increment:lst-ctn-kix_list_3-0}#doc18949057 ol.lst-kix_list_4-0.start{counter-reset:lst-ctn-kix_list_4-0 0}#doc18949057 .lst-kix_list_3-6>li{counter-increment:lst-ctn-kix_list_3-6}#doc18949057 .lst-kix_list_6-2>li:before{content:»» counter(lst-ctn-kix_list_6-2,lower-roman) «. «}#doc18949057 .lst-kix_list_2-5>li{counter-increment:lst-ctn-kix_list_2-5}#doc18949057 .lst-kix_list_2-8>li{counter-increment:lst-ctn-kix_list_2-8}#doc18949057 ol.lst-kix_list_3-2.start{counter-reset:lst-ctn-kix_list_3-2 0}#doc18949057 .lst-kix_list_6-8>li:before{content:»» counter(lst-ctn-kix_list_6-8,lower-roman) «. «}#doc18949057 .lst-kix_list_6-5>li:before{content:»» counter(lst-ctn-kix_list_6-5,lower-roman) «. «}#doc18949057 .lst-kix_list_6-7>li:before{content:»» counter(lst-ctn-kix_list_6-7,lower-latin) «. «}#doc18949057 ol.lst-kix_list_2-4.start{counter-reset:lst-ctn-kix_list_2-4 0}#doc18949057 .lst-kix_list_6-6>li:before{content:»» counter(lst-ctn-kix_list_6-6,decimal) «. «}#doc18949057 ol.lst-kix_list_1-3{list-style-type:none}#doc18949057 ol.lst-kix_list_1-4{list-style-type:none}#doc18949057 .lst-kix_list_2-7>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «.
lst-kix_list_6-4{list-style-type:none}#doc18949057 ol.lst-kix_list_6-5{list-style-type:none}#doc18949057 ol.lst-kix_list_1-0.start{counter-reset:lst-ctn-kix_list_1-0 0}#doc18949057 .lst-kix_list_6-1>li:before{content:»» counter(lst-ctn-kix_list_6-1,lower-latin) «. «}#doc18949057 .lst-kix_list_6-3>li:before{content:»» counter(lst-ctn-kix_list_6-3,decimal) «. «}#doc18949057 .lst-kix_list_6-5>li{counter-increment:lst-ctn-kix_list_6-5}#doc18949057 .lst-kix_list_6-8>li{counter-increment:lst-ctn-kix_list_6-8}#doc18949057 .lst-kix_list_6-0>li:before{content:»» counter(lst-ctn-kix_list_6-0,decimal) «. «}#doc18949057 .lst-kix_list_6-4>li:before{content:»» counter(lst-ctn-kix_list_6-4,lower-latin) «. «}#doc18949057 .lst-kix_list_3-0>li{counter-increment:lst-ctn-kix_list_3-0}#doc18949057 ol.lst-kix_list_4-0.start{counter-reset:lst-ctn-kix_list_4-0 0}#doc18949057 .lst-kix_list_3-6>li{counter-increment:lst-ctn-kix_list_3-6}#doc18949057 .lst-kix_list_6-2>li:before{content:»» counter(lst-ctn-kix_list_6-2,lower-roman) «. «}#doc18949057 .lst-kix_list_2-5>li{counter-increment:lst-ctn-kix_list_2-5}#doc18949057 .lst-kix_list_2-8>li{counter-increment:lst-ctn-kix_list_2-8}#doc18949057 ol.lst-kix_list_3-2.start{counter-reset:lst-ctn-kix_list_3-2 0}#doc18949057 .lst-kix_list_6-8>li:before{content:»» counter(lst-ctn-kix_list_6-8,lower-roman) «. «}#doc18949057 .lst-kix_list_6-5>li:before{content:»» counter(lst-ctn-kix_list_6-5,lower-roman) «. «}#doc18949057 .lst-kix_list_6-7>li:before{content:»» counter(lst-ctn-kix_list_6-7,lower-latin) «. «}#doc18949057 ol.lst-kix_list_2-4.start{counter-reset:lst-ctn-kix_list_2-4 0}#doc18949057 .lst-kix_list_6-6>li:before{content:»» counter(lst-ctn-kix_list_6-6,decimal) «. «}#doc18949057 ol.lst-kix_list_1-3{list-style-type:none}#doc18949057 ol.lst-kix_list_1-4{list-style-type:none}#doc18949057 .lst-kix_list_2-7>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «. » counter(lst-ctn-kix_list_2-4,decimal) «.» counter(lst-ctn-kix_list_2-5,decimal) «.» counter(lst-ctn-kix_list_2-6,decimal) «.» counter(lst-ctn-kix_list_2-7,decimal) » «}#doc18949057 .lst-kix_list_2-7>li{counter-increment:lst-ctn-kix_list_2-7}#doc18949057 ol.lst-kix_list_1-5{list-style-type:none}#doc18949057 .lst-kix_list_7-4>li:before{content:»» counter(lst-ctn-kix_list_7-4,lower-latin) «. «}#doc18949057 .lst-kix_list_7-6>li:before{content:»» counter(lst-ctn-kix_list_7-6,decimal) «. «}#doc18949057 ol.lst-kix_list_1-6{list-style-type:none}#doc18949057 ol.lst-kix_list_1-0{list-style-type:none}#doc18949057 .lst-kix_list_2-5>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «.» counter(lst-ctn-kix_list_2-4,decimal) «.» counter(lst-ctn-kix_list_2-5,decimal) » «}#doc18949057 ol.lst-kix_list_6-2.start{counter-reset:lst-ctn-kix_list_6-2 0}#doc18949057 ol.lst-kix_list_1-1{list-style-type:none}#doc18949057 ol.lst-kix_list_1-2{list-style-type:none}#doc18949057 .lst-kix_list_7-2>li:before{content:»» counter(lst-ctn-kix_list_7-2,lower-roman) «. «}#doc18949057 .lst-kix_list_7-6>li{counter-increment:lst-ctn-kix_list_7-6}#doc18949057 ol.lst-kix_list_4-6.start{counter-reset:lst-ctn-kix_list_4-6 0}#doc18949057 ol.lst-kix_list_3-0.start{counter-reset:lst-ctn-kix_list_3-0 0}#doc18949057 .lst-kix_list_5-7>li{counter-increment:lst-ctn-kix_list_5-7}#doc18949057 .lst-kix_list_7-7>li{counter-increment:lst-ctn-kix_list_7-7}#doc18949057 .lst-kix_list_7-8>li:before{content:»» counter(lst-ctn-kix_list_7-8,lower-roman) «. «}#doc18949057 ol.lst-kix_list_4-3.start{counter-reset:lst-ctn-kix_list_4-3 0}#doc18949057 ol.lst-kix_list_1-7{list-style-type:none}#doc18949057 .lst-kix_list_4-7>li{counter-increment:lst-ctn-kix_list_4-7}#doc18949057 ol.lst-kix_list_1-8{list-style-type:none}#doc18949057 .lst-kix_list_7-8>li{counter-increment:lst-ctn-kix_list_7-8}#doc18949057 ol.
» counter(lst-ctn-kix_list_2-4,decimal) «.» counter(lst-ctn-kix_list_2-5,decimal) «.» counter(lst-ctn-kix_list_2-6,decimal) «.» counter(lst-ctn-kix_list_2-7,decimal) » «}#doc18949057 .lst-kix_list_2-7>li{counter-increment:lst-ctn-kix_list_2-7}#doc18949057 ol.lst-kix_list_1-5{list-style-type:none}#doc18949057 .lst-kix_list_7-4>li:before{content:»» counter(lst-ctn-kix_list_7-4,lower-latin) «. «}#doc18949057 .lst-kix_list_7-6>li:before{content:»» counter(lst-ctn-kix_list_7-6,decimal) «. «}#doc18949057 ol.lst-kix_list_1-6{list-style-type:none}#doc18949057 ol.lst-kix_list_1-0{list-style-type:none}#doc18949057 .lst-kix_list_2-5>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «.» counter(lst-ctn-kix_list_2-4,decimal) «.» counter(lst-ctn-kix_list_2-5,decimal) » «}#doc18949057 ol.lst-kix_list_6-2.start{counter-reset:lst-ctn-kix_list_6-2 0}#doc18949057 ol.lst-kix_list_1-1{list-style-type:none}#doc18949057 ol.lst-kix_list_1-2{list-style-type:none}#doc18949057 .lst-kix_list_7-2>li:before{content:»» counter(lst-ctn-kix_list_7-2,lower-roman) «. «}#doc18949057 .lst-kix_list_7-6>li{counter-increment:lst-ctn-kix_list_7-6}#doc18949057 ol.lst-kix_list_4-6.start{counter-reset:lst-ctn-kix_list_4-6 0}#doc18949057 ol.lst-kix_list_3-0.start{counter-reset:lst-ctn-kix_list_3-0 0}#doc18949057 .lst-kix_list_5-7>li{counter-increment:lst-ctn-kix_list_5-7}#doc18949057 .lst-kix_list_7-7>li{counter-increment:lst-ctn-kix_list_7-7}#doc18949057 .lst-kix_list_7-8>li:before{content:»» counter(lst-ctn-kix_list_7-8,lower-roman) «. «}#doc18949057 ol.lst-kix_list_4-3.start{counter-reset:lst-ctn-kix_list_4-3 0}#doc18949057 ol.lst-kix_list_1-7{list-style-type:none}#doc18949057 .lst-kix_list_4-7>li{counter-increment:lst-ctn-kix_list_4-7}#doc18949057 ol.lst-kix_list_1-8{list-style-type:none}#doc18949057 .lst-kix_list_7-8>li{counter-increment:lst-ctn-kix_list_7-8}#doc18949057 ol. lst-kix_list_2-5.start{counter-reset:lst-ctn-kix_list_2-5 0}#doc18949057 .lst-kix_list_2-6>li{counter-increment:lst-ctn-kix_list_2-6}#doc18949057 .lst-kix_list_4-1>li:before{content:»» counter(lst-ctn-kix_list_4-1,lower-latin) «. «}#doc18949057 ol.lst-kix_list_7-3.start{counter-reset:lst-ctn-kix_list_7-3 0}#doc18949057 .lst-kix_list_4-3>li:before{content:»» counter(lst-ctn-kix_list_4-3,decimal) «. «}#doc18949057 .lst-kix_list_4-5>li:before{content:»» counter(lst-ctn-kix_list_4-5,lower-roman) «. «}#doc18949057 ol.lst-kix_list_5-7.start{counter-reset:lst-ctn-kix_list_5-7 0}#doc18949057 .lst-kix_list_1-8>li{counter-increment:lst-ctn-kix_list_1-8}#doc18949057 ol.lst-kix_list_1-4.start{counter-reset:lst-ctn-kix_list_1-4 0}#doc18949057 .lst-kix_list_5-5>li{counter-increment:lst-ctn-kix_list_5-5}#doc18949057 .lst-kix_list_3-5>li{counter-increment:lst-ctn-kix_list_3-5}#doc18949057 ol.lst-kix_list_1-1.start{counter-reset:lst-ctn-kix_list_1-1 0}#doc18949057 .lst-kix_list_3-4>li{counter-increment:lst-ctn-kix_list_3-4}#doc18949057 ol.lst-kix_list_4-4.start{counter-reset:lst-ctn-kix_list_4-4 0}#doc18949057 .lst-kix_list_6-4>li{counter-increment:lst-ctn-kix_list_6-4}#doc18949057 .lst-kix_list_6-3>li{counter-increment:lst-ctn-kix_list_6-3}#doc18949057 ol.lst-kix_list_1-3.start{counter-reset:lst-ctn-kix_list_1-3 0}#doc18949057 ol.lst-kix_list_2-8.start{counter-reset:lst-ctn-kix_list_2-8 0}#doc18949057 ol.lst-kix_list_1-2.start{counter-reset:lst-ctn-kix_list_1-2 0}#doc18949057 ol.lst-kix_list_7-6.start{counter-reset:lst-ctn-kix_list_7-6 0}#doc18949057 ol.lst-kix_list_6-1.start{counter-reset:lst-ctn-kix_list_6-1 0}#doc18949057 .lst-kix_list_1-1>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) » «}#doc18949057 .lst-kix_list_1-3>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) » «}#doc18949057 .lst-kix_list_4-8>li{counter-increment:lst-ctn-kix_list_4-8}#doc18949057 .
lst-kix_list_2-5.start{counter-reset:lst-ctn-kix_list_2-5 0}#doc18949057 .lst-kix_list_2-6>li{counter-increment:lst-ctn-kix_list_2-6}#doc18949057 .lst-kix_list_4-1>li:before{content:»» counter(lst-ctn-kix_list_4-1,lower-latin) «. «}#doc18949057 ol.lst-kix_list_7-3.start{counter-reset:lst-ctn-kix_list_7-3 0}#doc18949057 .lst-kix_list_4-3>li:before{content:»» counter(lst-ctn-kix_list_4-3,decimal) «. «}#doc18949057 .lst-kix_list_4-5>li:before{content:»» counter(lst-ctn-kix_list_4-5,lower-roman) «. «}#doc18949057 ol.lst-kix_list_5-7.start{counter-reset:lst-ctn-kix_list_5-7 0}#doc18949057 .lst-kix_list_1-8>li{counter-increment:lst-ctn-kix_list_1-8}#doc18949057 ol.lst-kix_list_1-4.start{counter-reset:lst-ctn-kix_list_1-4 0}#doc18949057 .lst-kix_list_5-5>li{counter-increment:lst-ctn-kix_list_5-5}#doc18949057 .lst-kix_list_3-5>li{counter-increment:lst-ctn-kix_list_3-5}#doc18949057 ol.lst-kix_list_1-1.start{counter-reset:lst-ctn-kix_list_1-1 0}#doc18949057 .lst-kix_list_3-4>li{counter-increment:lst-ctn-kix_list_3-4}#doc18949057 ol.lst-kix_list_4-4.start{counter-reset:lst-ctn-kix_list_4-4 0}#doc18949057 .lst-kix_list_6-4>li{counter-increment:lst-ctn-kix_list_6-4}#doc18949057 .lst-kix_list_6-3>li{counter-increment:lst-ctn-kix_list_6-3}#doc18949057 ol.lst-kix_list_1-3.start{counter-reset:lst-ctn-kix_list_1-3 0}#doc18949057 ol.lst-kix_list_2-8.start{counter-reset:lst-ctn-kix_list_2-8 0}#doc18949057 ol.lst-kix_list_1-2.start{counter-reset:lst-ctn-kix_list_1-2 0}#doc18949057 ol.lst-kix_list_7-6.start{counter-reset:lst-ctn-kix_list_7-6 0}#doc18949057 ol.lst-kix_list_6-1.start{counter-reset:lst-ctn-kix_list_6-1 0}#doc18949057 .lst-kix_list_1-1>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) » «}#doc18949057 .lst-kix_list_1-3>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) » «}#doc18949057 .lst-kix_list_4-8>li{counter-increment:lst-ctn-kix_list_4-8}#doc18949057 . lst-kix_list_1-7>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) «.» counter(lst-ctn-kix_list_1-5,decimal) «.» counter(lst-ctn-kix_list_1-6,decimal) «.» counter(lst-ctn-kix_list_1-7,decimal) » «}#doc18949057 ol.lst-kix_list_5-8.start{counter-reset:lst-ctn-kix_list_5-8 0}#doc18949057 ol.lst-kix_list_2-7.start{counter-reset:lst-ctn-kix_list_2-7 0}#doc18949057 .lst-kix_list_1-3>li{counter-increment:lst-ctn-kix_list_1-3}#doc18949057 .lst-kix_list_1-5>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) «.» counter(lst-ctn-kix_list_1-5,decimal) » «}#doc18949057 .lst-kix_list_5-6>li{counter-increment:lst-ctn-kix_list_5-6}#doc18949057 ol.lst-kix_list_7-5.start{counter-reset:lst-ctn-kix_list_7-5 0}#doc18949057 .lst-kix_list_2-1>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) » «}#doc18949057 ol.lst-kix_list_6-0.start{counter-reset:lst-ctn-kix_list_6-0 0}#doc18949057 .lst-kix_list_2-3>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) » «}#doc18949057 .lst-kix_list_4-2>li{counter-increment:lst-ctn-kix_list_4-2}#doc18949057 ol.lst-kix_list_3-1{list-style-type:none}#doc18949057 ol.lst-kix_list_3-2{list-style-type:none}#doc18949057 .lst-kix_list_3-1>li{counter-increment:lst-ctn-kix_list_3-1}#doc18949057 ol.lst-kix_list_3-3{list-style-type:none}#doc18949057 ol.lst-kix_list_3-4.start{counter-reset:lst-ctn-kix_list_3-4 0}#doc18949057 .lst-kix_list_5-1>li{counter-increment:lst-ctn-kix_list_5-1}#doc18949057 ol.lst-kix_list_3-4{list-style-type:none}#doc18949057 ol.
lst-kix_list_1-7>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) «.» counter(lst-ctn-kix_list_1-5,decimal) «.» counter(lst-ctn-kix_list_1-6,decimal) «.» counter(lst-ctn-kix_list_1-7,decimal) » «}#doc18949057 ol.lst-kix_list_5-8.start{counter-reset:lst-ctn-kix_list_5-8 0}#doc18949057 ol.lst-kix_list_2-7.start{counter-reset:lst-ctn-kix_list_2-7 0}#doc18949057 .lst-kix_list_1-3>li{counter-increment:lst-ctn-kix_list_1-3}#doc18949057 .lst-kix_list_1-5>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) «.» counter(lst-ctn-kix_list_1-5,decimal) » «}#doc18949057 .lst-kix_list_5-6>li{counter-increment:lst-ctn-kix_list_5-6}#doc18949057 ol.lst-kix_list_7-5.start{counter-reset:lst-ctn-kix_list_7-5 0}#doc18949057 .lst-kix_list_2-1>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) » «}#doc18949057 ol.lst-kix_list_6-0.start{counter-reset:lst-ctn-kix_list_6-0 0}#doc18949057 .lst-kix_list_2-3>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) » «}#doc18949057 .lst-kix_list_4-2>li{counter-increment:lst-ctn-kix_list_4-2}#doc18949057 ol.lst-kix_list_3-1{list-style-type:none}#doc18949057 ol.lst-kix_list_3-2{list-style-type:none}#doc18949057 .lst-kix_list_3-1>li{counter-increment:lst-ctn-kix_list_3-1}#doc18949057 ol.lst-kix_list_3-3{list-style-type:none}#doc18949057 ol.lst-kix_list_3-4.start{counter-reset:lst-ctn-kix_list_3-4 0}#doc18949057 .lst-kix_list_5-1>li{counter-increment:lst-ctn-kix_list_5-1}#doc18949057 ol.lst-kix_list_3-4{list-style-type:none}#doc18949057 ol. lst-kix_list_3-0{list-style-type:none}#doc18949057 .lst-kix_list_1-1>li{counter-increment:lst-ctn-kix_list_1-1}#doc18949057 .lst-kix_list_7-1>li{counter-increment:lst-ctn-kix_list_7-1}#doc18949057 ol.lst-kix_list_2-6.start{counter-reset:lst-ctn-kix_list_2-6 0}#doc18949057 .lst-kix_list_3-0>li:before{content:»» counter(lst-ctn-kix_list_3-0,decimal) «. «}#doc18949057 ol.lst-kix_list_7-7.start{counter-reset:lst-ctn-kix_list_7-7 0}#doc18949057 .lst-kix_list_3-1>li:before{content:»» counter(lst-ctn-kix_list_3-1,lower-latin) «. «}#doc18949057 .lst-kix_list_3-2>li:before{content:»» counter(lst-ctn-kix_list_3-2,lower-roman) «. «}#doc18949057 ol.lst-kix_list_1-8.start{counter-reset:lst-ctn-kix_list_1-8 0}#doc18949057 .lst-kix_list_4-0>li{counter-increment:lst-ctn-kix_list_4-0}#doc18949057 .lst-kix_list_6-0>li{counter-increment:lst-ctn-kix_list_6-0}#doc18949057 .lst-kix_list_3-5>li:before{content:»» counter(lst-ctn-kix_list_3-5,lower-roman) «. «}#doc18949057 .lst-kix_list_3-4>li:before{content:»» counter(lst-ctn-kix_list_3-4,lower-latin) «. «}#doc18949057 .lst-kix_list_3-3>li:before{content:»» counter(lst-ctn-kix_list_3-3,decimal) «. «}#doc18949057 ol.lst-kix_list_3-5{list-style-type:none}#doc18949057 ol.lst-kix_list_3-6{list-style-type:none}#doc18949057 ol.lst-kix_list_3-7{list-style-type:none}#doc18949057 ol.lst-kix_list_3-8{list-style-type:none}#doc18949057 .lst-kix_list_3-8>li:before{content:»» counter(lst-ctn-kix_list_3-8,lower-roman) «. «}#doc18949057 .lst-kix_list_2-0>li{counter-increment:lst-ctn-kix_list_2-0}#doc18949057 .lst-kix_list_3-6>li:before{content:»» counter(lst-ctn-kix_list_3-6,decimal) «. «}#doc18949057 .lst-kix_list_3-7>li:before{content:»» counter(lst-ctn-kix_list_3-7,lower-latin) «. «}#doc18949057 ol.lst-kix_list_5-0.start{counter-reset:lst-ctn-kix_list_5-0 0}#doc18949057 ol.lst-kix_list_4-2.start{counter-reset:lst-ctn-kix_list_4-2 0}#doc18949057 ol.lst-kix_list_2-2{list-style-type:none}#doc18949057 ol.lst-kix_list_2-3{list-style-type:none}#doc18949057 ol.
lst-kix_list_3-0{list-style-type:none}#doc18949057 .lst-kix_list_1-1>li{counter-increment:lst-ctn-kix_list_1-1}#doc18949057 .lst-kix_list_7-1>li{counter-increment:lst-ctn-kix_list_7-1}#doc18949057 ol.lst-kix_list_2-6.start{counter-reset:lst-ctn-kix_list_2-6 0}#doc18949057 .lst-kix_list_3-0>li:before{content:»» counter(lst-ctn-kix_list_3-0,decimal) «. «}#doc18949057 ol.lst-kix_list_7-7.start{counter-reset:lst-ctn-kix_list_7-7 0}#doc18949057 .lst-kix_list_3-1>li:before{content:»» counter(lst-ctn-kix_list_3-1,lower-latin) «. «}#doc18949057 .lst-kix_list_3-2>li:before{content:»» counter(lst-ctn-kix_list_3-2,lower-roman) «. «}#doc18949057 ol.lst-kix_list_1-8.start{counter-reset:lst-ctn-kix_list_1-8 0}#doc18949057 .lst-kix_list_4-0>li{counter-increment:lst-ctn-kix_list_4-0}#doc18949057 .lst-kix_list_6-0>li{counter-increment:lst-ctn-kix_list_6-0}#doc18949057 .lst-kix_list_3-5>li:before{content:»» counter(lst-ctn-kix_list_3-5,lower-roman) «. «}#doc18949057 .lst-kix_list_3-4>li:before{content:»» counter(lst-ctn-kix_list_3-4,lower-latin) «. «}#doc18949057 .lst-kix_list_3-3>li:before{content:»» counter(lst-ctn-kix_list_3-3,decimal) «. «}#doc18949057 ol.lst-kix_list_3-5{list-style-type:none}#doc18949057 ol.lst-kix_list_3-6{list-style-type:none}#doc18949057 ol.lst-kix_list_3-7{list-style-type:none}#doc18949057 ol.lst-kix_list_3-8{list-style-type:none}#doc18949057 .lst-kix_list_3-8>li:before{content:»» counter(lst-ctn-kix_list_3-8,lower-roman) «. «}#doc18949057 .lst-kix_list_2-0>li{counter-increment:lst-ctn-kix_list_2-0}#doc18949057 .lst-kix_list_3-6>li:before{content:»» counter(lst-ctn-kix_list_3-6,decimal) «. «}#doc18949057 .lst-kix_list_3-7>li:before{content:»» counter(lst-ctn-kix_list_3-7,lower-latin) «. «}#doc18949057 ol.lst-kix_list_5-0.start{counter-reset:lst-ctn-kix_list_5-0 0}#doc18949057 ol.lst-kix_list_4-2.start{counter-reset:lst-ctn-kix_list_4-2 0}#doc18949057 ol.lst-kix_list_2-2{list-style-type:none}#doc18949057 ol.lst-kix_list_2-3{list-style-type:none}#doc18949057 ol. lst-kix_list_2-4{list-style-type:none}#doc18949057 ol.lst-kix_list_7-2.start{counter-reset:lst-ctn-kix_list_7-2 0}#doc18949057 ol.lst-kix_list_2-5{list-style-type:none}#doc18949057 .lst-kix_list_4-4>li{counter-increment:lst-ctn-kix_list_4-4}#doc18949057 ol.lst-kix_list_2-0{list-style-type:none}#doc18949057 ol.lst-kix_list_2-1{list-style-type:none}#doc18949057 .lst-kix_list_4-8>li:before{content:»» counter(lst-ctn-kix_list_4-8,lower-roman) «. «}#doc18949057 ol.lst-kix_list_6-4.start{counter-reset:lst-ctn-kix_list_6-4 0}#doc18949057 .lst-kix_list_4-7>li:before{content:»» counter(lst-ctn-kix_list_4-7,lower-latin) «. «}#doc18949057 ol.lst-kix_list_5-6.start{counter-reset:lst-ctn-kix_list_5-6 0}#doc18949057 ol.lst-kix_list_4-1.start{counter-reset:lst-ctn-kix_list_4-1 0}#doc18949057 .lst-kix_list_7-3>li{counter-increment:lst-ctn-kix_list_7-3}#doc18949057 ol.lst-kix_list_4-8.start{counter-reset:lst-ctn-kix_list_4-8 0}#doc18949057 ol.lst-kix_list_3-3.start{counter-reset:lst-ctn-kix_list_3-3 0}#doc18949057 ol.lst-kix_list_2-6{list-style-type:none}#doc18949057 ol.lst-kix_list_2-7{list-style-type:none}#doc18949057 ol.lst-kix_list_2-8{list-style-type:none}#doc18949057 ol.lst-kix_list_7-8.start{counter-reset:lst-ctn-kix_list_7-8 0}#doc18949057 ol.lst-kix_list_7-1.start{counter-reset:lst-ctn-kix_list_7-1 0}#doc18949057 .lst-kix_list_3-3>li{counter-increment:lst-ctn-kix_list_3-3}#doc18949057 ol.lst-kix_list_6-3.start{counter-reset:lst-ctn-kix_list_6-3 0}#doc18949057 ol.lst-kix_list_5-5.start{counter-reset:lst-ctn-kix_list_5-5 0}#doc18949057 .lst-kix_list_7-0>li:before{content:»» counter(lst-ctn-kix_list_7-0,decimal) «. «}#doc18949057 .lst-kix_list_2-2>li{counter-increment:lst-ctn-kix_list_2-2}#doc18949057 ol.lst-kix_list_4-7.start{counter-reset:lst-ctn-kix_list_4-7 0}#doc18949057 .lst-kix_list_6-2>li{counter-increment:lst-ctn-kix_list_6-2}#doc18949057 ol.lst-kix_list_5-0{list-style-type:none}#doc18949057 .lst-kix_list_2-6>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.
lst-kix_list_2-4{list-style-type:none}#doc18949057 ol.lst-kix_list_7-2.start{counter-reset:lst-ctn-kix_list_7-2 0}#doc18949057 ol.lst-kix_list_2-5{list-style-type:none}#doc18949057 .lst-kix_list_4-4>li{counter-increment:lst-ctn-kix_list_4-4}#doc18949057 ol.lst-kix_list_2-0{list-style-type:none}#doc18949057 ol.lst-kix_list_2-1{list-style-type:none}#doc18949057 .lst-kix_list_4-8>li:before{content:»» counter(lst-ctn-kix_list_4-8,lower-roman) «. «}#doc18949057 ol.lst-kix_list_6-4.start{counter-reset:lst-ctn-kix_list_6-4 0}#doc18949057 .lst-kix_list_4-7>li:before{content:»» counter(lst-ctn-kix_list_4-7,lower-latin) «. «}#doc18949057 ol.lst-kix_list_5-6.start{counter-reset:lst-ctn-kix_list_5-6 0}#doc18949057 ol.lst-kix_list_4-1.start{counter-reset:lst-ctn-kix_list_4-1 0}#doc18949057 .lst-kix_list_7-3>li{counter-increment:lst-ctn-kix_list_7-3}#doc18949057 ol.lst-kix_list_4-8.start{counter-reset:lst-ctn-kix_list_4-8 0}#doc18949057 ol.lst-kix_list_3-3.start{counter-reset:lst-ctn-kix_list_3-3 0}#doc18949057 ol.lst-kix_list_2-6{list-style-type:none}#doc18949057 ol.lst-kix_list_2-7{list-style-type:none}#doc18949057 ol.lst-kix_list_2-8{list-style-type:none}#doc18949057 ol.lst-kix_list_7-8.start{counter-reset:lst-ctn-kix_list_7-8 0}#doc18949057 ol.lst-kix_list_7-1.start{counter-reset:lst-ctn-kix_list_7-1 0}#doc18949057 .lst-kix_list_3-3>li{counter-increment:lst-ctn-kix_list_3-3}#doc18949057 ol.lst-kix_list_6-3.start{counter-reset:lst-ctn-kix_list_6-3 0}#doc18949057 ol.lst-kix_list_5-5.start{counter-reset:lst-ctn-kix_list_5-5 0}#doc18949057 .lst-kix_list_7-0>li:before{content:»» counter(lst-ctn-kix_list_7-0,decimal) «. «}#doc18949057 .lst-kix_list_2-2>li{counter-increment:lst-ctn-kix_list_2-2}#doc18949057 ol.lst-kix_list_4-7.start{counter-reset:lst-ctn-kix_list_4-7 0}#doc18949057 .lst-kix_list_6-2>li{counter-increment:lst-ctn-kix_list_6-2}#doc18949057 ol.lst-kix_list_5-0{list-style-type:none}#doc18949057 .lst-kix_list_2-6>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «. » counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «.» counter(lst-ctn-kix_list_2-4,decimal) «.» counter(lst-ctn-kix_list_2-5,decimal) «.» counter(lst-ctn-kix_list_2-6,decimal) » «}#doc18949057 .lst-kix_list_3-7>li{counter-increment:lst-ctn-kix_list_3-7}#doc18949057 ol.lst-kix_list_5-1{list-style-type:none}#doc18949057 ol.lst-kix_list_5-2{list-style-type:none}#doc18949057 .lst-kix_list_2-4>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «.» counter(lst-ctn-kix_list_2-4,decimal) » «}#doc18949057 .lst-kix_list_2-8>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «.» counter(lst-ctn-kix_list_2-4,decimal) «.» counter(lst-ctn-kix_list_2-5,decimal) «.» counter(lst-ctn-kix_list_2-6,decimal) «.» counter(lst-ctn-kix_list_2-7,decimal) «.» counter(lst-ctn-kix_list_2-8,decimal) » «}#doc18949057 .lst-kix_list_7-1>li:before{content:»» counter(lst-ctn-kix_list_7-1,lower-latin) «. «}#doc18949057 .lst-kix_list_7-5>li:before{content:»» counter(lst-ctn-kix_list_7-5,lower-roman) «. «}#doc18949057 .lst-kix_list_6-6>li{counter-increment:lst-ctn-kix_list_6-6}#doc18949057 ol.lst-kix_list_5-4.start{counter-reset:lst-ctn-kix_list_5-4 0}#doc18949057 .lst-kix_list_7-3>li:before{content:»» counter(lst-ctn-kix_list_7-3,decimal) «. «}#doc18949057 ol.lst-kix_list_5-1.start{counter-reset:lst-ctn-kix_list_5-1 0}#doc18949057 ol.lst-kix_list_5-7{list-style-type:none}#doc18949057 .lst-kix_list_6-7>li{counter-increment:lst-ctn-kix_list_6-7}#doc18949057 ol.lst-kix_list_5-8{list-style-type:none}#doc18949057 ol.lst-kix_list_5-3{list-style-type:none}#doc18949057 ol.lst-kix_list_5-4{list-style-type:none}#doc18949057 .lst-kix_list_1-7>li{counter-increment:lst-ctn-kix_list_1-7}#doc18949057 ol.
» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «.» counter(lst-ctn-kix_list_2-4,decimal) «.» counter(lst-ctn-kix_list_2-5,decimal) «.» counter(lst-ctn-kix_list_2-6,decimal) » «}#doc18949057 .lst-kix_list_3-7>li{counter-increment:lst-ctn-kix_list_3-7}#doc18949057 ol.lst-kix_list_5-1{list-style-type:none}#doc18949057 ol.lst-kix_list_5-2{list-style-type:none}#doc18949057 .lst-kix_list_2-4>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «.» counter(lst-ctn-kix_list_2-4,decimal) » «}#doc18949057 .lst-kix_list_2-8>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) «.» counter(lst-ctn-kix_list_2-3,decimal) «.» counter(lst-ctn-kix_list_2-4,decimal) «.» counter(lst-ctn-kix_list_2-5,decimal) «.» counter(lst-ctn-kix_list_2-6,decimal) «.» counter(lst-ctn-kix_list_2-7,decimal) «.» counter(lst-ctn-kix_list_2-8,decimal) » «}#doc18949057 .lst-kix_list_7-1>li:before{content:»» counter(lst-ctn-kix_list_7-1,lower-latin) «. «}#doc18949057 .lst-kix_list_7-5>li:before{content:»» counter(lst-ctn-kix_list_7-5,lower-roman) «. «}#doc18949057 .lst-kix_list_6-6>li{counter-increment:lst-ctn-kix_list_6-6}#doc18949057 ol.lst-kix_list_5-4.start{counter-reset:lst-ctn-kix_list_5-4 0}#doc18949057 .lst-kix_list_7-3>li:before{content:»» counter(lst-ctn-kix_list_7-3,decimal) «. «}#doc18949057 ol.lst-kix_list_5-1.start{counter-reset:lst-ctn-kix_list_5-1 0}#doc18949057 ol.lst-kix_list_5-7{list-style-type:none}#doc18949057 .lst-kix_list_6-7>li{counter-increment:lst-ctn-kix_list_6-7}#doc18949057 ol.lst-kix_list_5-8{list-style-type:none}#doc18949057 ol.lst-kix_list_5-3{list-style-type:none}#doc18949057 ol.lst-kix_list_5-4{list-style-type:none}#doc18949057 .lst-kix_list_1-7>li{counter-increment:lst-ctn-kix_list_1-7}#doc18949057 ol. lst-kix_list_3-8.start{counter-reset:lst-ctn-kix_list_3-8 0}#doc18949057 ol.lst-kix_list_5-5{list-style-type:none}#doc18949057 ol.lst-kix_list_5-6{list-style-type:none}#doc18949057 .lst-kix_list_7-7>li:before{content:»» counter(lst-ctn-kix_list_7-7,lower-latin) «. «}#doc18949057 .lst-kix_list_7-5>li{counter-increment:lst-ctn-kix_list_7-5}#doc18949057 .lst-kix_list_5-8>li{counter-increment:lst-ctn-kix_list_5-8}#doc18949057 .lst-kix_list_4-0>li:before{content:»» counter(lst-ctn-kix_list_4-0,decimal) » «}#doc18949057 .lst-kix_list_3-8>li{counter-increment:lst-ctn-kix_list_3-8}#doc18949057 ol.lst-kix_list_6-8.start{counter-reset:lst-ctn-kix_list_6-8 0}#doc18949057 .lst-kix_list_4-6>li{counter-increment:lst-ctn-kix_list_4-6}#doc18949057 ol.lst-kix_list_1-7.start{counter-reset:lst-ctn-kix_list_1-7 0}#doc18949057 .lst-kix_list_4-4>li:before{content:»» counter(lst-ctn-kix_list_4-4,lower-latin) «. «}#doc18949057 ol.lst-kix_list_2-2.start{counter-reset:lst-ctn-kix_list_2-2 0}#doc18949057 .lst-kix_list_1-5>li{counter-increment:lst-ctn-kix_list_1-5}#doc18949057 ol.lst-kix_list_6-5.start{counter-reset:lst-ctn-kix_list_6-5 0}#doc18949057 .lst-kix_list_4-2>li:before{content:»» counter(lst-ctn-kix_list_4-2,lower-roman) «. «}#doc18949057 .lst-kix_list_4-6>li:before{content:»» counter(lst-ctn-kix_list_4-6,decimal) «. «}#doc18949057 ol.lst-kix_list_7-0.start{counter-reset:lst-ctn-kix_list_7-0 0}#doc18949057 ol.lst-kix_list_4-0{list-style-type:none}#doc18949057 ol.lst-kix_list_4-1{list-style-type:none}#doc18949057 ol.lst-kix_list_4-2{list-style-type:none}#doc18949057 ol.lst-kix_list_4-3{list-style-type:none}#doc18949057 .lst-kix_list_2-4>li{counter-increment:lst-ctn-kix_list_2-4}#doc18949057 ol.lst-kix_list_6-7.start{counter-reset:lst-ctn-kix_list_6-7 0}#doc18949057 ol.lst-kix_list_3-6.start{counter-reset:lst-ctn-kix_list_3-6 0}#doc18949057 .lst-kix_list_5-3>li{counter-increment:lst-ctn-kix_list_5-3}#doc18949057 ol.lst-kix_list_4-8{list-style-type:none}#doc18949057 .lst-kix_list_7-4>li{counter-increment:lst-ctn-kix_list_7-4}#doc18949057 .
lst-kix_list_3-8.start{counter-reset:lst-ctn-kix_list_3-8 0}#doc18949057 ol.lst-kix_list_5-5{list-style-type:none}#doc18949057 ol.lst-kix_list_5-6{list-style-type:none}#doc18949057 .lst-kix_list_7-7>li:before{content:»» counter(lst-ctn-kix_list_7-7,lower-latin) «. «}#doc18949057 .lst-kix_list_7-5>li{counter-increment:lst-ctn-kix_list_7-5}#doc18949057 .lst-kix_list_5-8>li{counter-increment:lst-ctn-kix_list_5-8}#doc18949057 .lst-kix_list_4-0>li:before{content:»» counter(lst-ctn-kix_list_4-0,decimal) » «}#doc18949057 .lst-kix_list_3-8>li{counter-increment:lst-ctn-kix_list_3-8}#doc18949057 ol.lst-kix_list_6-8.start{counter-reset:lst-ctn-kix_list_6-8 0}#doc18949057 .lst-kix_list_4-6>li{counter-increment:lst-ctn-kix_list_4-6}#doc18949057 ol.lst-kix_list_1-7.start{counter-reset:lst-ctn-kix_list_1-7 0}#doc18949057 .lst-kix_list_4-4>li:before{content:»» counter(lst-ctn-kix_list_4-4,lower-latin) «. «}#doc18949057 ol.lst-kix_list_2-2.start{counter-reset:lst-ctn-kix_list_2-2 0}#doc18949057 .lst-kix_list_1-5>li{counter-increment:lst-ctn-kix_list_1-5}#doc18949057 ol.lst-kix_list_6-5.start{counter-reset:lst-ctn-kix_list_6-5 0}#doc18949057 .lst-kix_list_4-2>li:before{content:»» counter(lst-ctn-kix_list_4-2,lower-roman) «. «}#doc18949057 .lst-kix_list_4-6>li:before{content:»» counter(lst-ctn-kix_list_4-6,decimal) «. «}#doc18949057 ol.lst-kix_list_7-0.start{counter-reset:lst-ctn-kix_list_7-0 0}#doc18949057 ol.lst-kix_list_4-0{list-style-type:none}#doc18949057 ol.lst-kix_list_4-1{list-style-type:none}#doc18949057 ol.lst-kix_list_4-2{list-style-type:none}#doc18949057 ol.lst-kix_list_4-3{list-style-type:none}#doc18949057 .lst-kix_list_2-4>li{counter-increment:lst-ctn-kix_list_2-4}#doc18949057 ol.lst-kix_list_6-7.start{counter-reset:lst-ctn-kix_list_6-7 0}#doc18949057 ol.lst-kix_list_3-6.start{counter-reset:lst-ctn-kix_list_3-6 0}#doc18949057 .lst-kix_list_5-3>li{counter-increment:lst-ctn-kix_list_5-3}#doc18949057 ol.lst-kix_list_4-8{list-style-type:none}#doc18949057 .lst-kix_list_7-4>li{counter-increment:lst-ctn-kix_list_7-4}#doc18949057 . lst-kix_list_1-0>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «. «}#doc18949057 ol.lst-kix_list_4-4{list-style-type:none}#doc18949057 ol.lst-kix_list_4-5{list-style-type:none}#doc18949057 .lst-kix_list_1-2>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) » «}#doc18949057 ol.lst-kix_list_2-0.start{counter-reset:lst-ctn-kix_list_2-0 0}#doc18949057 ol.lst-kix_list_4-6{list-style-type:none}#doc18949057 ol.lst-kix_list_4-7{list-style-type:none}#doc18949057 .lst-kix_list_1-4>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) » «}#doc18949057 ol.lst-kix_list_3-5.start{counter-reset:lst-ctn-kix_list_3-5 0}#doc18949057 .lst-kix_list_1-0>li{counter-increment:lst-ctn-kix_list_1-0}#doc18949057 .lst-kix_list_1-6>li{counter-increment:lst-ctn-kix_list_1-6}#doc18949057 .lst-kix_list_1-6>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) «.» counter(lst-ctn-kix_list_1-5,decimal) «.» counter(lst-ctn-kix_list_1-6,decimal) » «}#doc18949057 .lst-kix_list_2-0>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «. «}#doc18949057 ol.lst-kix_list_2-1.start{counter-reset:lst-ctn-kix_list_2-1 0}#doc18949057 .lst-kix_list_4-5>li{counter-increment:lst-ctn-kix_list_4-5}#doc18949057 .lst-kix_list_1-8>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) «.» counter(lst-ctn-kix_list_1-5,decimal) «.» counter(lst-ctn-kix_list_1-6,decimal) «.» counter(lst-ctn-kix_list_1-7,decimal) «.
lst-kix_list_1-0>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «. «}#doc18949057 ol.lst-kix_list_4-4{list-style-type:none}#doc18949057 ol.lst-kix_list_4-5{list-style-type:none}#doc18949057 .lst-kix_list_1-2>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) » «}#doc18949057 ol.lst-kix_list_2-0.start{counter-reset:lst-ctn-kix_list_2-0 0}#doc18949057 ol.lst-kix_list_4-6{list-style-type:none}#doc18949057 ol.lst-kix_list_4-7{list-style-type:none}#doc18949057 .lst-kix_list_1-4>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) » «}#doc18949057 ol.lst-kix_list_3-5.start{counter-reset:lst-ctn-kix_list_3-5 0}#doc18949057 .lst-kix_list_1-0>li{counter-increment:lst-ctn-kix_list_1-0}#doc18949057 .lst-kix_list_1-6>li{counter-increment:lst-ctn-kix_list_1-6}#doc18949057 .lst-kix_list_1-6>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) «.» counter(lst-ctn-kix_list_1-5,decimal) «.» counter(lst-ctn-kix_list_1-6,decimal) » «}#doc18949057 .lst-kix_list_2-0>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «. «}#doc18949057 ol.lst-kix_list_2-1.start{counter-reset:lst-ctn-kix_list_2-1 0}#doc18949057 .lst-kix_list_4-5>li{counter-increment:lst-ctn-kix_list_4-5}#doc18949057 .lst-kix_list_1-8>li:before{content:»» counter(lst-ctn-kix_list_1-0,decimal) «.» counter(lst-ctn-kix_list_1-1,decimal) «.» counter(lst-ctn-kix_list_1-2,decimal) «.» counter(lst-ctn-kix_list_1-3,decimal) «.» counter(lst-ctn-kix_list_1-4,decimal) «.» counter(lst-ctn-kix_list_1-5,decimal) «.» counter(lst-ctn-kix_list_1-6,decimal) «.» counter(lst-ctn-kix_list_1-7,decimal) «. » counter(lst-ctn-kix_list_1-8,decimal) » «}#doc18949057 .lst-kix_list_2-2>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) » «}#doc18949057 ol.lst-kix_list_5-2.start{counter-reset:lst-ctn-kix_list_5-2 0}#doc18949057 ol{margin:0;padding:0}#doc18949057 table td,table th{padding:0}#doc18949057 .c1{padding-top:0pt;text-indent:35.5pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:justify;height:12pt}#doc18949057 .c34{-webkit-text-decoration-skip:none;color:#0000ff;font-weight:400;text-decoration:underline;vertical-align:baseline;text-decoration-skip-ink:none;font-family:»Times New Roman»;font-style:normal}#doc18949057 .c16{padding-top:14pt;text-indent:35.5pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:center;height:12pt}#doc18949057 .c0{color:#000000;font-weight:400;text-decoration:none;vertical-align:baseline;font-size:14pt;font-family:»Times New Roman»;font-style:normal}#doc18949057 .c6{padding-top:0pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:center;height:12pt}#doc18949057 .c3{padding-top:14pt;text-indent:35.5pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c14{color:#000000;font-weight:400;text-decoration:none;vertical-align:baseline;font-size:28pt;font-family:»Times New Roman»;font-style:normal}#doc18949057 .c36{padding-top:14pt;text-indent:35.5pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:center}#doc18949057 .c2{padding-top:0pt;text-indent:35.5pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c4{margin-left:35.5pt;padding-top:14pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c35{padding-top:14pt;text-indent:35.5pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:center}#doc18949057 .c5{padding-top:0pt;padding-bottom:0pt;line-height:1.0;orphans:2;widows:2;text-align:center;height:12pt}#doc18949057 .
» counter(lst-ctn-kix_list_1-8,decimal) » «}#doc18949057 .lst-kix_list_2-2>li:before{content:»» counter(lst-ctn-kix_list_2-0,decimal) «.» counter(lst-ctn-kix_list_2-1,decimal) «.» counter(lst-ctn-kix_list_2-2,decimal) » «}#doc18949057 ol.lst-kix_list_5-2.start{counter-reset:lst-ctn-kix_list_5-2 0}#doc18949057 ol{margin:0;padding:0}#doc18949057 table td,table th{padding:0}#doc18949057 .c1{padding-top:0pt;text-indent:35.5pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:justify;height:12pt}#doc18949057 .c34{-webkit-text-decoration-skip:none;color:#0000ff;font-weight:400;text-decoration:underline;vertical-align:baseline;text-decoration-skip-ink:none;font-family:»Times New Roman»;font-style:normal}#doc18949057 .c16{padding-top:14pt;text-indent:35.5pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:center;height:12pt}#doc18949057 .c0{color:#000000;font-weight:400;text-decoration:none;vertical-align:baseline;font-size:14pt;font-family:»Times New Roman»;font-style:normal}#doc18949057 .c6{padding-top:0pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:center;height:12pt}#doc18949057 .c3{padding-top:14pt;text-indent:35.5pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c14{color:#000000;font-weight:400;text-decoration:none;vertical-align:baseline;font-size:28pt;font-family:»Times New Roman»;font-style:normal}#doc18949057 .c36{padding-top:14pt;text-indent:35.5pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:center}#doc18949057 .c2{padding-top:0pt;text-indent:35.5pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c4{margin-left:35.5pt;padding-top:14pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c35{padding-top:14pt;text-indent:35.5pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:center}#doc18949057 .c5{padding-top:0pt;padding-bottom:0pt;line-height:1.0;orphans:2;widows:2;text-align:center;height:12pt}#doc18949057 . c9{padding-top:0pt;padding-bottom:0pt;line-height:1.0;orphans:2;widows:2;text-align:left;height:12pt}#doc18949057 .c25{padding-top:14pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c17{padding-top:0pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c12{padding-top:0pt;padding-bottom:0pt;line-height:1.0;orphans:2;widows:2;text-align:center}#doc18949057 .c38{padding-top:0pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:left}#doc18949057 .c37{padding-top:0pt;padding-bottom:0pt;line-height:1.0;orphans:2;widows:2;text-align:left}#doc18949057 .c11{padding-top:0pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:center}#doc18949057 .c10{font-weight:700;text-decoration:none;vertical-align:baseline;font-family:»Times New Roman»;font-style:normal}#doc18949057 .c31{margin-left:71.5pt;padding-left:18pt;margin-right:2.9pt}#doc18949057 .c18{margin-left:28.4pt;text-indent:-28.4pt;height:12pt}#doc18949057 .c23{margin-left:53.5pt;padding-left:0pt;margin-right:2.9pt}#doc18949057 .c21{margin-left:53.5pt;text-indent:-36pt;margin-right:2.9pt}#doc18949057 .c33{max-width:467.7pt;padding:56.7pt 42.5pt 56.7pt 85pt}#doc18949057 .c24{margin-left:28.4pt;text-indent:7pt}#doc18949057 .c22{margin-left:-21.2pt;text-indent:21.2pt}#doc18949057 .c27{margin-left:0pt;list-style-position:inside}#doc18949057 .c7{color:inherit;text-decoration:inherit}#doc18949057 .c20{margin-left:-21.3pt;text-indent:21.3pt}#doc18949057 .c28{margin-left:36pt;padding-left:0pt}#doc18949057 .c8{color:#000000;font-size:14pt}#doc18949057 .c26{padding:0;margin:0}#doc18949057 .c29{text-indent:28.4pt;height:12pt}#doc18949057 .c40{font-weight:400}#doc18949057 .c32{text-decoration:none}#doc18949057 .c13{background-color:#ffffff}#doc18949057 .c39{margin-right:2.9pt}#doc18949057 .c19{font-weight:700}#doc18949057 .c15{font-size:14pt}#doc18949057 .c30{height:12pt}#doc18949057 .title{padding-top:24pt;color:#000000;font-weight:700;font-size:36pt;padding-bottom:6pt;font-family:»Times New Roman»;line-height:1.
c9{padding-top:0pt;padding-bottom:0pt;line-height:1.0;orphans:2;widows:2;text-align:left;height:12pt}#doc18949057 .c25{padding-top:14pt;padding-bottom:14pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c17{padding-top:0pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:justify}#doc18949057 .c12{padding-top:0pt;padding-bottom:0pt;line-height:1.0;orphans:2;widows:2;text-align:center}#doc18949057 .c38{padding-top:0pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:left}#doc18949057 .c37{padding-top:0pt;padding-bottom:0pt;line-height:1.0;orphans:2;widows:2;text-align:left}#doc18949057 .c11{padding-top:0pt;padding-bottom:0pt;line-height:1.5;orphans:2;widows:2;text-align:center}#doc18949057 .c10{font-weight:700;text-decoration:none;vertical-align:baseline;font-family:»Times New Roman»;font-style:normal}#doc18949057 .c31{margin-left:71.5pt;padding-left:18pt;margin-right:2.9pt}#doc18949057 .c18{margin-left:28.4pt;text-indent:-28.4pt;height:12pt}#doc18949057 .c23{margin-left:53.5pt;padding-left:0pt;margin-right:2.9pt}#doc18949057 .c21{margin-left:53.5pt;text-indent:-36pt;margin-right:2.9pt}#doc18949057 .c33{max-width:467.7pt;padding:56.7pt 42.5pt 56.7pt 85pt}#doc18949057 .c24{margin-left:28.4pt;text-indent:7pt}#doc18949057 .c22{margin-left:-21.2pt;text-indent:21.2pt}#doc18949057 .c27{margin-left:0pt;list-style-position:inside}#doc18949057 .c7{color:inherit;text-decoration:inherit}#doc18949057 .c20{margin-left:-21.3pt;text-indent:21.3pt}#doc18949057 .c28{margin-left:36pt;padding-left:0pt}#doc18949057 .c8{color:#000000;font-size:14pt}#doc18949057 .c26{padding:0;margin:0}#doc18949057 .c29{text-indent:28.4pt;height:12pt}#doc18949057 .c40{font-weight:400}#doc18949057 .c32{text-decoration:none}#doc18949057 .c13{background-color:#ffffff}#doc18949057 .c39{margin-right:2.9pt}#doc18949057 .c19{font-weight:700}#doc18949057 .c15{font-size:14pt}#doc18949057 .c30{height:12pt}#doc18949057 .title{padding-top:24pt;color:#000000;font-weight:700;font-size:36pt;padding-bottom:6pt;font-family:»Times New Roman»;line-height:1. 0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 .subtitle{padding-top:18pt;color:#666666;font-size:24pt;padding-bottom:4pt;font-family:»Georgia»;line-height:1.0;page-break-after:avoid;font-style:italic;orphans:2;widows:2;text-align:left}#doc18949057 li{color:#000000;font-size:12pt;font-family:»Times New Roman»}#doc18949057 p{margin:0;color:#000000;font-size:12pt;font-family:»Times New Roman»}#doc18949057 h2{padding-top:24pt;color:#000000;font-weight:700;font-size:24pt;padding-bottom:6pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h3{padding-top:18pt;color:#000000;font-weight:700;font-size:18pt;padding-bottom:4pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h4{padding-top:14pt;color:#000000;font-weight:700;font-size:14pt;padding-bottom:4pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h5{padding-top:12pt;color:#000000;font-weight:700;font-size:12pt;padding-bottom:2pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h5{padding-top:11pt;color:#000000;font-weight:700;font-size:11pt;padding-bottom:2pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h6{padding-top:10pt;color:#000000;font-weight:700;font-size:10pt;padding-bottom:2pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 ]]>
0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 .subtitle{padding-top:18pt;color:#666666;font-size:24pt;padding-bottom:4pt;font-family:»Georgia»;line-height:1.0;page-break-after:avoid;font-style:italic;orphans:2;widows:2;text-align:left}#doc18949057 li{color:#000000;font-size:12pt;font-family:»Times New Roman»}#doc18949057 p{margin:0;color:#000000;font-size:12pt;font-family:»Times New Roman»}#doc18949057 h2{padding-top:24pt;color:#000000;font-weight:700;font-size:24pt;padding-bottom:6pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h3{padding-top:18pt;color:#000000;font-weight:700;font-size:18pt;padding-bottom:4pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h4{padding-top:14pt;color:#000000;font-weight:700;font-size:14pt;padding-bottom:4pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h5{padding-top:12pt;color:#000000;font-weight:700;font-size:12pt;padding-bottom:2pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h5{padding-top:11pt;color:#000000;font-weight:700;font-size:11pt;padding-bottom:2pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 h6{padding-top:10pt;color:#000000;font-weight:700;font-size:10pt;padding-bottom:2pt;font-family:»Times New Roman»;line-height:1.0;page-break-after:avoid;orphans:2;widows:2;text-align:left}#doc18949057 ]]> А.
А.